


Abstract
Background: Familial hypercholesterolemia (FH) is associated with premature atherosclerotic cardiovascular disease caused by elevated low-density lipoprotein cholesterol (LDL-C) levels. We determined the impact of a full next-generation sequencing (NGS) genetic panel on reclassification of patients with a clinical diagnosis of FH in Quebec compared to the partial genetic panel currently offered by the Quebec Ministère de la Santé et des Services sociaux (Ministry of Health and Social Services) (MSSS), which includes 11 variants that are common in French Canadians.
Methods: We conducted a retrospective cohort study in a subgroup of patients in the Canadian FH Registry seen at the McGill University Health Centre Preventive Cardiology/Lipid Clinic, Montréal, between September 2017 and September 2021 who were clinically diagnosed with severe hypercholesterolemia, probable FH or definite FH according to the Canadian definition of FH. Next-generation sequencing of the LDLR, APOB and PCSK9 genes, and multiplex ligation-dependent probe amplification of the LDLR gene to detect genetic variants, were performed.
Results: Among 335 consecutive patients with heterozygous FH (184 men [54.9%] and 151 women [45.1%]), the baseline LDL-C level was 6.96 (standard deviation 1.79) mmol/L. Patients identified through cascade screening were 11 years younger on average than index patients, and smaller proportions presented to the clinic with cardiovascular risk factors. A pathogenic FH variant was identified in 169 (73.8%) of the 229 patients who underwent genetic testing; the majority had variants in the LDLR (146 [86.4%]) or APOB (24 [14.2%]) gene. The genetic panel offered by the MSSS accounted for only 48% of the variants identified with the full NGS panel. Of the 229 patients, 90 (39.3%, 95% confidence interval 32.9%–46.0%) were reclassified from a clinical diagnosis of probable FH to definite FH after genetic screening with a full FH panel.
Interpretation: Genetic testing in patients suspected of having FH provided diagnostic certainty and permitted many patients with a clinical diagnosis of probable FH to be reclassified as having definite FH. Genetic screening allows for increased identification of patients with FH and may therefore help reduce the burden of cardiovascular disease and mortality rates among Canadians with FH. Trial registration: ClinicalTrials.gov, no. NCT02009345
Heterozygous familial hypercholesterolemia (FH) is the most common autosomal genetic disorder, with a prevalence of 1/311 worldwide.1 In some regions of Quebec, Canada, with a historically isolated population with a high rate of endogamy and little genetic mixing (a founder effect), such as Kamouraska, Côte-Nord and Saguenay–Lac-St-Jean, the prevalence has been reported to be as high as 1/80.2 Familial hypercholesterolemia is most commonly caused by variants in the LDLR, APOB or PCSK9 gene, resulting in markedly elevated plasma levels of low-density lipoprotein cholesterol (LDL-C) and premature atherosclerotic cardiovascular disease (CVD).3,4 Untreated men with FH develop clinical atherosclerotic CVD by their mid-30s, and women, by their mid-40s.5 Prompt recognition of FH and initiation of lipid-lowering therapy is highly efficacious and can markedly reduce patients’ atherosclerotic CVD risk to that of an age-matched population.5,6 Despite this, FH remains underdiagnosed and undertreated, with less than 15% of cases identified in Canada.5 Methods to improve diagnosis are needed. The disorder can be diagnosed with clinical criteria algorithms, such as the Simon Broome Criteria,7 the Dutch Lipid Clinic Network criteria8 or the new Canadian FH definition,9 which consider patients’ LDL-C level, the presence of tendon xanthomas and family history, or, increasingly, by means of genetic testing. In countries such as the Netherlands and Norway, genetic testing is offered to nearly all people in whom FH is suspected.10 In addition to helping detect FH, knowledge of a person’s genotype can facilitate cascade screening of family members and enable further assessment of their CVD risk. Khera and colleagues11 showed that the presence of an FH variant increases CVD risk, irrespective of LDL-C levels. The type of FH variant is equally important, as null or negative variants impose a twofold higher CVD risk compared to milder hypomorphic or defective variants.12
Although genetic testing is not necessarily required for the diagnosis of FH, it is nevertheless considered the gold standard and is recommended by several professional organizations, including the Canadian Cardiovascular Society, the US Centers for Disease Control and Prevention Office of Genomics and Precision Public Health, the International Atherosclerosis Society and the United Kingdom National Institute for Health and Care Excellence.13–16 Despite these recommendations, genetic testing for FH is not routinely available as part of clinical care in Canada, which may contribute to the low diagnosis rates in this country.5,10
A few academic medical centres in Canada perform complete DNA analysis of the main genes causing FH, but these are on a research basis. In the province of Quebec, the Ministère de la Santé et des Services sociaux (Ministry of Health and Social Services) (MSSS) offers clinically certified genetic screening for the 11 most commonly known French-Canadian variants in LDLR, but the APOB and PCSK9 genes are not included. To address this, we established a genetic screening program of the LDLR, APOB and PCSK9 genes in Canada. The objective of the present study was to examine the impact of unbiased full next-generation sequencing (NGS) on reclassification of patients with a clinical diagnosis of FH in Quebec based on the Canadian definition of FH compared to the partial genetic panel covering only French-Canadian variants currently offered by the MSSS.
Methods
Study design and setting
We conducted a retrospective cohort study among patients seen in the Preventive Cardiology/Lipid Clinic of the McGill University Health Centre at the Royal Victoria Hospital in Montréal from September 2017 to September 2021, corresponding to the development period of the genetic test for FH at the site. The catchment area for the health centre includes the Montréal metropolitan area and the Réseau universitaire intégré de Santé et Services Sociaux McGill, covering an area spanning 63% of the territory of Quebec and comprising 7 regional health authorities (https://www.mcgill.ca/ruisss/territory). At the time of data collection (at the registration visit), the McGill University Health Centre Preventive Cardiology/Lipid Clinic was 1 of 19 main academic Canadian FH Registry participating sites and 1 of 4 registry sites in the province (Appendix 1, available at www.cmajopen.ca/content/11/4/E754/suppl/DC1). Biochemical and DNA samples were collected as planned in the Canadian FH Registry protocol; details of the registry have been previously published.17 The study reporting followed the recommendations of the Reporting of Studies Conducted using Observational Routinely Collected Health Data (RECORD) statement (http://record-statement.org/).
Participants
Only patients with a clinical diagnosis of FH seen at the McGill University Health Centre registry site were included in the present study. People were recruited into the Canadian FH Registry if they were referred to the clinic for an LDL-C level higher than the 95th percentile for age and sex (the currently accepted diagnostic cut-point for FH9) or from cascade screening of family members of an index patient previously seen at the clinic. To be included in the present study, participants had to be adult (age ≥ 18 yr), have been seen in the McGill University Health Centre Preventive Cardiology/Lipid Clinic, have a clinical diagnosis of definite FH, probable FH or severe hypercholesterolemia according to the Canadian definition of FH9 (main study cohort), and have received a result of genetic testing for FH (genetic testing outcome subgroup).
Data sources
Clinical data
The clinical data were extracted from the Canadian FH Registry17 database by the national coordinator (I.R.) in September 2021. Briefly, data on patients’ demographic characteristics, medical history and medication, family history of premature CVD and dyslipidemia, physical signs of FH such as tendon xanthomas and untreated lipid profile were obtained by a cardiologist with expertise in FH (J.G.). Secondary causes of high LDL-C levels were ruled out.9 A standard nonfasting blood sample was collected, and plasma levels of total cholesterol, high-density lipoprotein cholesterol (HDL-C), triglycerides, apolipoprotein B, lipoprotein, thyroid-stimulating hormone, and hepatic transaminase, creatinine and creatine kinase were measured by standard automated assays performed by the OptiLab Montréal–McGill University Health Centre biochemistry laboratory. The LDL-C level was calculated with the Friedewald formula. When untreated LDL-C levels were unavailable, we imputed them based on the LDL-C levels with the patient receiving treatment, and the actual dosage and type of lipid-lowering therapy used at the time of analysis, as previously described.18
The initial FH diagnosis was established for each patient according to the Canadian definition of FH,9 first using only clinical criteria data, and then with the addition of genetic testing results (reclassification). Briefly, in the Canadian definition, the high LDL-C cut-off points are 5.0 mmol/L or higher for those aged 40 years or older, 4.5 mmol/L or higher for those aged 18–39 years, and 4.0 mmol/L or higher for those younger than 18 years. Patients are classified as having definite FH if they have high untreated LDL-C levels combined with a causal DNA variant (LDLR, APOB or PCSK9) or tendon xanthomas, or if they have an untreated LDL-C level of 8.5 mmol/L or higher. Patients are diagnosed as having probable FH if they have high LDL-C levels and a first-degree relative with high LDL-C levels or premature atherosclerotic CVD (atherosclerotic CVD event before age 55 for men and 65 for women13). Patients are diagnosed as having severe hypercholesterolemia if the only diagnostic criterion they present with is a high LDL-C level.9
Genetic testing
Genetic testing for FH was done specifically for the purpose of the present study. Next-generation sequencing of the LDLR, PCSK9 and APOB genes, and multiplex ligation-dependent probe amplification19 for detection of copy number variants in the LDLR gene were carried out at the Core Molecular Diagnostic Laboratory of the McGill University Health Centre. This laboratory is currently the only clinical molecular genetics laboratory for FH in Canada certified by the Clinical Laboratory Improvement Amendments of 1988 regulations (certification by the US Food and Drug Administration, the US Centers for Medicare & Medicaid Services and the Centers for Disease Control and Prevention). Briefly, all coding bases and splice junctions of the LDLR, APOB and PCSK9 genes were amplified at a sequencing depth of at least 20× with the use of custom multiplex polymerase chain reaction tests. In cases of ambiguity, Sanger sequencing was used to confirm variants detected by NGS.
Standard bioinformatics software and databases were used for data analysis, from management of raw sequencing data to clinical annotation of identified variants. We interpreted DNA variants as per the 2015 American College of Medical Genetics and the Association for Molecular Pathology guidelines for variant prioritization20 with the use of several databases, including dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), the Human Gene Mutation Database, the Leiden Open Variation Database (LOVD) 3.0 (https://databases.lovd.nl/shared/genes), the Western Database of Lipid Variants21 and the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/). Briefly, when examining variants in LDLR, APOB or PCSK9 genes, we took into consideration whether they had previously been reported as pathogenic, likely pathogenic or a variant of unknown significance. We applied a systematic approach by considering the ClinVar significance, allele frequency databases and disease association. We arbitrarily considered main FH variants to be those identified in more than 1.5% of cases.
We compared the list of FH variants that we identified in the cohort using our new clinical genetic screening protocol to the list of French-Canadian variants currently available from the provincially approved genetic assay. Currently, the MSSS reimburses genetic screening for specific variants of the LDLR gene only, which are variants commonly seen in the French-Canadian population: 2 copy number variants (delta 5 Kb, delta > 15 Kb) and 9 single-nucleotide variants (Trp66Gly, Cys646Tyr, Glu207Lys, Cys152Trp, Arg329Xaa, Cys347Arg, Tyr468Xaa, Tyr354Cys, 681ins7) (https://www.msss.gouv.qc.ca/repertoires/biomed/index.php).
Statistical analysis
We presented patient demographic characteristics and lipid profiles using standard descriptive statistics, including mean and standard deviation (SD), median and interquartile range, and frequency with percentage. We used statistical testing to compare demographic characteristics and lipid profiles between index patients and patients identified by cascade screening, and between males and females, to assess for differences between these key subgroups. We used the Student t test to compare continuous variables unless they had a skewed distribution (triglycerides and lipoprotein(a)), in which case we used the Mann–Whitney test. We used the χ2 test to compare categoric variables. The initial clinical diagnosis of FH was made using the Canadian definition for FH,9 and the same definition was applied to patients after genetic testing for FH from both methods used (i.e., partial MSSS FH panel or full NGS panel). We compared individual classifications of the updated clinical diagnosis after the partial MSSS FH panel with those obtained after the full NGS genetic panel. We calculated 95% confidence intervals (CIs) for the proportion of patients diagnosed as having probable FH who were reclassified as having definite FH using a binomial distribution. Significance was set at p < 0.05. We used SPSS Statistics 24 (IBM Corp.) for all analyses.
Ethics approval
The study was approved by the Research Ethics Board of the McGill University Health Centre (REB no. 13-292-BMD), and all patients signed informed consent forms for data collection and genetic analysis. The Canadian FH Registry is registered at www.ClinicalTrials.gov (NCT02009345).
Results
The cohort in this retrospective analysis consisted of 335 consecutive participants with heterozygous FH (184 men [54.9%], mean age at registration 48 [SD 14] yr, and 151 women [45.1%], mean age at registration 52 [SD 17] yr) (Figure 1). The mean age at the time of registration of the overall cohort was 50 (SD 15) years. All patients had a diagnosis of severe hypercholesterolemia, probable FH or definite FH according to the Canadian definition of FH.9 The major baseline characteristics of the cohort are presented in Table 1. Of the 335 patients, 75 (22.4%) had hypertension, 97 (29.0%) had coronary artery disease (CAD), and 80 (23.9%) presented with tendon xanthomas. A majority of the patients (257/314 [81.8%]) were of European descent, with 172/314 (54.8%) self-identifying as French Canadian. At the time of registration, the mean LDL-C level was 3.61 (SD 2.05) mmol/L, and 255/335 patients (76.1%) were receiving lipid-lowering therapy. However, the mean recorded baseline LDL-C level was 6.96 (SD 1.79) mmol/L.
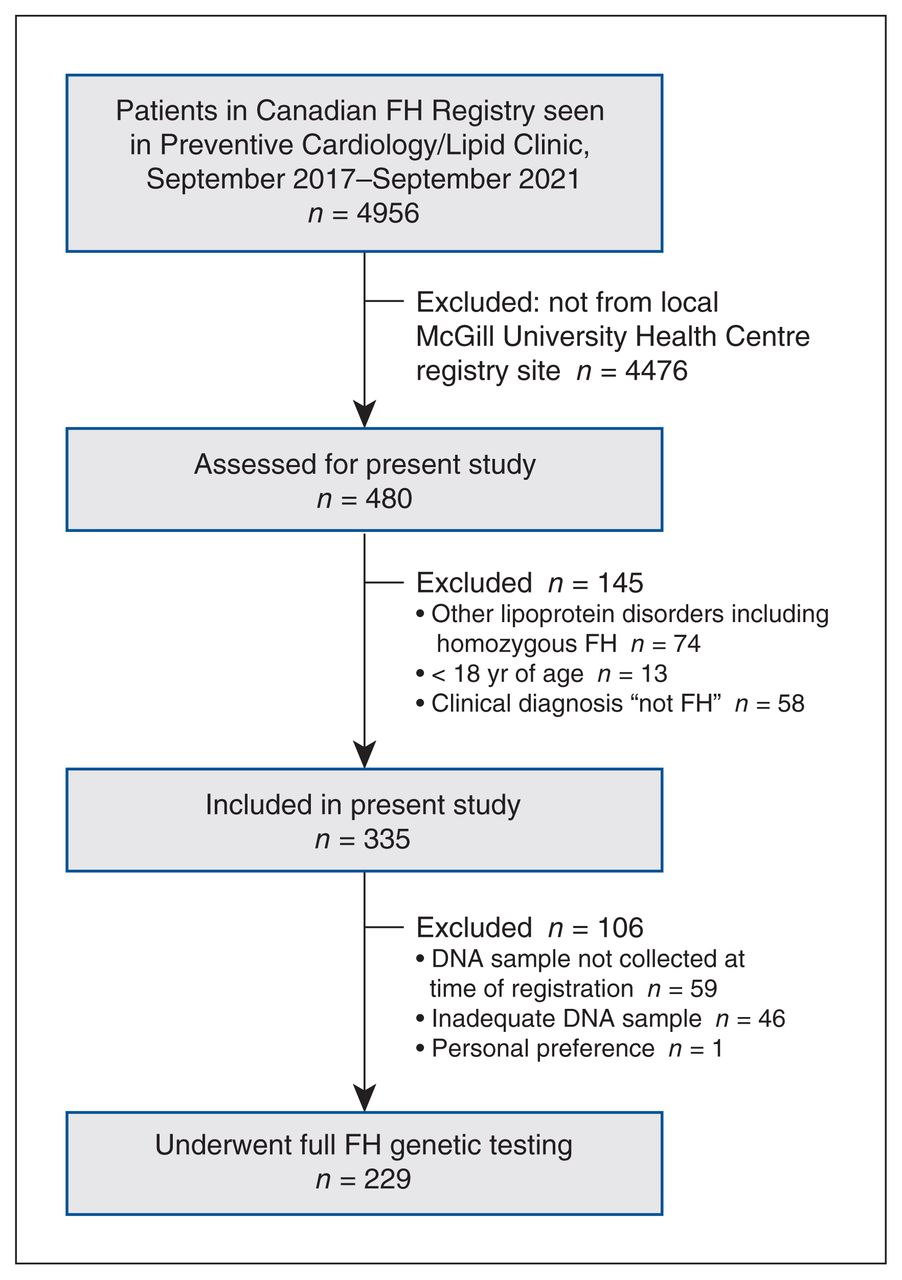
Flow diagram showing patient selection. Note: FH = familial hypercholesterolemia.
Characteristics of patients with heterozygous familial hypercholesterolemia, September 2017 to September 2021
Of the 335 patients, 288 (157 men [54.5%] with mean age at registration 51 [SD 15] yr) were index patients, and 47 (27 men [57.4%] with mean age at registration 41 [SD 15] yr) were identified through cascade screening (no large families included) (Table 1). At the time of diagnosis of FH, index patients were 11 years older on average than those identified through cascade screening (41 yr v. 30 yr, p < 0.001), and were more likely to have a history of hypertension (71 [24.6%] v. 4 [8.5%], p = 0.01) and CAD (93 [32.3%] v. 4 [8.5%], p < 0.001). At registration, more index patients than those identified through cascade screening were receiving lipid-lowering therapy (223 [77.4%] v. 32 [68.1%]); however, the difference was not statistically significant (p = 0.2). Index patients had significantly lower mean levels of LDL-C (3.51 [SD 2.02] mmol/L v. 4.27 [SD 2.15] mmol/L, p = 0.02) and apolipoprotein B (1.16 [SD 0.46] g/L v. 1.34 [SD 0.57] g/L, p = 0.02) than those identified through cascade screening. Index patients had significantly higher baseline untreated triglyceride levels at first diagnosis than those identified through cascade screening (p = 0.009).
Men were diagnosed 6 years earlier on average than women (37 yr v. 43 yr, p = 0.001) and were more likely to have a history of CAD (68 [37.0%] v. 29 [19.2%], p < 0.001) (Table 2). At registration, a higher proportion of men than women were receiving lipid-lowering therapy (155 [84.2%] v. 100 [66.2%]). Interestingly, men had significantly lower levels of total cholesterol, LDL-C, HDL-C and apolipoprotein B than women at registration (p < 0.001 for all). However, men and women had similar lipid levels at first diagnosis, with the exception that men had a higher mean triglyceride level (1.75 [SD 1.21] mmol/L v. 1.49 [SD 1.37] mmol/L, p = 0.02) and lower mean HDL-C level (1.16 [SD 0.26] mmol/L v. 1.44 [SD 0.39] mmol/L, p < 0.001).
Characteristics of patients by sex
Genetic testing
Full FH genetic testing was done in 229 patients (68.4%) (189 in the index group and 40 in the cascade screening group), and an FH variant was identified in 169 (73.8%) (132 and 37, respectively), including a patient with an APOE variant identified after additional genetic analysis. The majority of patients with a positive result were found to have variants within the LDLR gene (146 [86.4%]), the APOB gene (24 [14.2%]) or the PCSK9 gene (6 [3.6%]); 11 patients had variants on more than 1 gene: 2 patients had multiple APOB variants, 2 patients had multiple LDLR variants, and 9 patients had a mix of LDLR, APOB and/or PCSK9 variants (Table 3). The LDLR delta 15 Kb, known as the “French-Canadian” mutation, was the most prevalent variant (55 patients [32.5%]). The second most prevalent variant was the LDLR p.Cys681* (15 [8.9%]), predominantly found in patients who were descendants of Christian Lebanese.22 The MSSS genetic panel accounted for only 48% of the variants identified with the full NGS panel. Even among the 92 patients with an FH variant who identified as French Canadian, 15 (16%) did not have a common variant listed in the panel. Similar proportions and types of variants were found in the patients identified via cascade screening as in index patients. Notably, a higher proportion of women than men had a PCSK9 variant (5/74 women [6.8%] v. 1/95 men [1.0%], p = 0.04) (Appendix 2, Supplemental Table S1, available at www.cmajopen.ca/content/11/4/E754/suppl/DC1); no other significant differences in variant prevalence were found between men and women. Table 3 shows the FH variants identified with a frequency greater than 1.5% in our cohort as well as with the MSSS genetic panel, and Appendix 2, Supplemental Table S2 lists all the variants identified, including variants of uncertain significance and newly identified variants. In total, 69 unique variants were identified in this cohort of patients (Appendix 2, Supplemental Table S2).
Main familial hypercholesterolemia variants and Quebec MSSS French-Canadian variants identified in the cohort
Familial hypercholesterolemia diagnosis
Table 4 and Table 5 show the number of patients classified as having a severe hypercholesterolemia, probable FH or definite FH phenotype based solely on clinical criteria and their reclassification after genetic testing results were considered, respectively. Before genetic testing, a majority of patients (134 [58.5%]) were diagnosed as having probable FH. Of the 229 patients who underwent genetic testing, more were reclassified from having probable FH to having definite FH after a full genetic testing panel than after MSSS partial genetic testing (90 [39.3%, 95% CI 32.9–46.0] v. 36 [15.7%, 95% CI 11.3 –21.1], p < 0.001, χ2 test). In 7 patients classified as having definite FH, no mutation in the LDLR, APOB or PCSK9 gene was identified (Appendix 2, Supplemental Table S3); further sequencing at the Robarts Research Institute, London, Ontario, Canada, and the Broad Institute, Cambridge, Massachusetts, identified mutations in the ABCG5/8 or APOE gene in 3 of these patients.
Classification of clinical diagnosis of familial hypercholesterolemia before genetic testing
Reclassification of clinical diagnosis of familial hypercholesterolemia after genetic testing
Interpretation
In our cohort of patients with FH in Quebec, 74% of all patients tested with our genetic screening protocol were found to have a genetic variant known to cause FH, well in keeping with data showing that about 20% of patients with a presumed diagnosis of FH may have a polygenic form of the disorder.23 The protocol allowed for the majority of patients clinically diagnosed as having probable FH to be reclassified as having definite FH. Most of the variants identified in our cohort were in the LDLR, APOB and PCSK9 genes, half of which were not covered by the MSSS genetic panel.
It is well known that FH is underdiagnosed and, thus, undertreated. It is estimated that more than 27 600–34 400 people in Quebec have FH,1 and data from the Canadian FH Registry show that less than 10% of these patients have thus far been identified.17 To further facilitate diagnosis, the Canadian FH definition,9 based on simplified clinical criteria or genetic testing for variants in the LDLR, APOB or PCSK9 genes with subsequent cascade screening to identify affected relatives effectively, was recently implemented in Canada. Previous studies have shown that genetic screening is highly effective in identifying patients and improving follow-up rates,5,12,24 and is considered the standard for the diagnosis of FH.25 The MSSS genetic panel covers screening of only 11 variants in LDLR commonly identified in French Canadians. This panel was originally developed to identify variants causing FH in patients living in regions with a founder effect, such as Kamouraska, Côte-Nord and Saguenay–Lac-St-Jean. Patients currently living in these regions are being referred mainly to the lipid clinic at the CHU de Québec–Université Laval. Many patients with FH whose parents and grandparents were from these regions have migrated to large cities such as Montréal and Québec, and made their way to the McGill University Health Centre Preventive Cardiology/Lipid Clinic. However, the MSSS genetic panel accounted for only 48% of variants identified with the full NGS panel in our cohort, and less than half of patients with a positive genetic result had variants listed in the MSSS panel. Only 32% of patients with a positive genetic test result were found to have the LDLR 15 Kb delta variant (known as the French-Canadian variant), a lower proportion than previously described.26
The search for variants only in the LDLR gene is not enough, considering that 18% of variants identified in the present study were on the APOB and PCSK9 genes. Improving identification of genetic variants in patients with FH will also allow for better assessment of their CVD risk and a potential change in treatment, even in patients already classified as having definite FH. Khera and colleagues11 reported that, regardless of LDL-C levels, patients with a confirmed pathogenetic FH variant have an elevated risk of CAD, and Lee and colleagues12 found that loss-of-function variants were associated with a twofold higher CAD risk compared to hypomorphic variants. Therefore, it is essential that patients with a presumptive clinical diagnosis of FH undergo complete unbiased genetic screening for FH in the LDLR, PCSK9 and APOB genes.
Where genetic screening may be most useful is in cases in which the clinical diagnosis of probable FH or severe hypercholesterolemia is made. A patient’s clinical diagnosis is often incomplete owing to the absence of specific diagnostic criteria such as untreated LDL-C, family history of CVD, dyslipidemia or xanthomas.9 Our genetic screening protocol allowed for a statistically significant improvement in identification and reclassification of patients with FH, with one-third of those classified as having severe hypercholesteremia and the majority of those classified as having probable FH being reclassified as having definite FH. This more accurate diagnosis may improve quality of care and compliance with treatment, encourage cascade screening and facilitate access to new drugs (e.g., PCSK9 inhibitors [evolocumab and alirocumab]) whose high cost may result in limited use.
Studies have shown that targeted cascade screening with DNA analysis is highly effective in identifying people with FH.24,27 In our cohort, patients identified through cascade screening were diagnosed considerably earlier than index patients and consequently presented to the lipid clinic in a healthier state: they had lower rates of hypertension and CAD, and lower baseline untreated triglyceride levels. These findings are in keeping with a previous study showing the importance of early diagnosis of FH for normal life expectancy.25 Umans-Eckenhausen and colleagues24 reported that, once identified through cascade screening, most affected people seek treatment and are successfully started on cholesterol-lowering treatment to lower their risk of premature CVD early. Ideally, cascade screening should be systematic and coordinated in specialized centres, and patients should be offered genetic counselling and long-term follow-up.5
Although no large families were included in the present study, more than 1 family member of index patients with the p.Ala431Thr and p.Asp90Asn variants were examined, which explains the observed differences in the prevalence of these variants between the index and cascade screening groups. No FH variant was identified in a few patients in the latter group, which meant they did not carry the FH family variant identified in their corresponding index patient. Nevertheless, their clinically severe hypercholesterolemia status — which may have been caused by other factors — was detected and treated according to guidelines.
We previously reported a possible sex difference in the treatment of women with FH.28 Our data and those from a large cohort study29 suggest that this is an underrecognized problem. Although minimal differences in the prevalence of variants were found between men and women in the present study, the number of patients examined was small. Therefore, the possibility of a relation between differences in variant types and sex disparities in the presentation and treatment of women with FH should be investigated further.
The genetic testing program for FH that we describe is accessible across Canada through the Canadian FH Registry (https://www.fhcanada.net/). Since the creation of the registry, in 2014, participants of the FH Canada network have worked together to publish new guidelines for the treatment of FH in Canada14,30 and a new validated definition of FH,9 and have created clinical tools for accurate diagnosis.9
Limitations
The molecular diagnosis of FH was limited to sequencing of the exome (the portion of genes that codes for the mature protein) of the LDLR, APOB and PCSK9 genes; thus, rare variants in APOE (e.g., p.Leu167del), LDL-R adaptor protein 1 (LDLRAP1), lysosomal acid lipase (LIPA) and the ATP binding cassette transporter G5 and G8 genotypes (ABCG5, ABCG8), which can cause a phenocopy of FH, will have been missed. In addition, because our genetic sequencing did not cover deep intronic portions of the LDLR, APOB or PCSK9 gene, these intronic variants (the portion of genes between exons that does not code for a protein but might be important in gene expression) will also have been missed. Second, it is known that some patients with elevated LDL-C levels do not have a monogenic variant in the genes known to cause FH but, rather, exhibit a cumulative sum of genetic polymorphisms in different genes increasing LDL-C levels in Mendelian randomization studies.23 Accordingly, Talmud and colleagues23 derived a “gene LDL-C score” in order to distinguish patients with polygenic and monogenic FH. Although our study did not focus on obtaining an LDL-C score, our findings are consistent with data showing that 20% of patients with a presumed diagnosis of FH may have a polygenic form of the disease.23 The statistical tests used assumed independence of observations; however, cascade sampling of family members leads to nonindependence, and our analyses were not clustered by index cases. In addition, the comparison of index cases to nonindex cases was limited by low statistical power. Last, our results are limited to a single-centre cohort from the McGill University Health Centre and therefore need validation in other centres or multicentre cohorts.
Conclusion
Our results suggest that sequencing of the LDLR, APOB and PCSK9 genes in patients suspected of having FH provides diagnostic certainty and valuable diagnostic reclassification. Ultimately, genetic diagnosis would allow for improved cascade screening. These results also have implications for health policies, such as the use of the genetic panel offered by the MSSS. As such, full unbiased genetic screening will allow for increased identification of patients with FH and help reduce the burden of CVD and mortality rates among Canadians with FH.
Footnotes
Competing interests: Jacques Genest is a consultant for Amgen, Novartis, Sanofi and Novo Nordisk. No other competing interests were declared.
This article has been peer reviewed.
Contributors: Jacques Genest conceived of and designed the study. Isabelle Ruel and Linda Fri Ngufor acquired the data, Amanda Guerin, Iulia Iatan and Isabelle Ruel analyzed the data, and Amanda Guerin, Iulia Iatan, Isabelle Ruel and Jacques Genest interpreted the data. Amanda Guerin and Iulia Iatan drafted the manuscript. All of the authors revised the manuscript critically for important intellectual content, approved the final version to be published and agreed to act as guarantors of the work.
Funding: Funding was obtained from unrestricted grants from Amgen and Sanofi Canada, as well as the Fonds de recherche du Québec – Santé/Réseau de recherche sur la santé cardiométabolique, le diabète et l’obésité and a Canadian Institutes of Health Research/FH Canada grant (PJT 168886).
Data sharing: The data that support the findings of this study are available from the corresponding author on reasonable request.
Supplemental information: For reviewer comments and the original submission of this manuscript, please see www.cmajopen.ca/content/11/4/E754/suppl/DC1.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
References
- © 2023 CMA Impact Inc. or its licensors
In this issue

{kind=link}
Article tools





Related Articles
Cited By...
- No citing articles found.