


Abstract
Background: The number of patients with sickle cell disease in Ontario, Canada, is unknown. In the absence of a formal registry, we performed a study to determine an approximate census via analysis of health administrative databases.
Methods: We identified Ontario patients with a diagnosis of sickle cell disease through queries of the Discharge Abstract Database, National Ambulatory Care Reporting System and Newborn Screening Ontario database. The period of inquiry was Apr. 1, 2007, through Mar. 31, 2017. We identified repeat interactions by the same patient by cross-referencing provincial health insurance plan numbers.
Results: We documented health care system interactions for 3418 unique patients (1912 [55.9%] female, median age at the time of identification 24 yr). Over the 10-year study period, patients visited the emergency department a median of 2 (interquartile range [IQR] 1–7) times and an average of 6.69 (standard deviation [SD] 26.71) times, and were admitted to hospital a median of 1 (IQR 1–5) time and an average of 4.38 (SD 8.53) times for treatment related to sickle cell disease. A total of 229 patients (6.7%) died during the study period, with an average age at death of 55 years. Even without accounting for the effects of immigration, the rate of natural increase slowed slightly over the study period owing to a decrease in the annual number of affected births.
Interpretation: The estimated prevalence of patients with sickle cell disease in Ontario in 2007/08–2016/17 was 1 in 4200, and affected patients’ need for hospital-based care was substantial, although highly variable. Similar queries of health administrative databases may be feasible in other Canadian provinces.
Sickle cell disease is an autosomal recessive condition and the most common single-gene disease worldwide,1 affecting about 7 million people.2 A total of 300 000–400 000 affected children are born each year.1 The most common genotypes are homozygous hemoglobin S, and compound heterozygosity of hemoglobin S with β-thalassemia (Hgb-S/β thal) or hemoglobin C.3 Although sickle cell disease was originally confined to areas of the world with endemic malaria, a growing proportion of affected people are now found in higher-resource countries of the Northern Hemisphere.4 In these settings, the provision of simple interventions such as prophylactic antibiotic treatment, hydroxyurea therapy and blood transfusion have resulted in dramatic increases in life expectancy, which now approaches 60 years.5 However, affected people still experience substantial morbidity owing to intermittent vaso-occlusive and hemolytic crises, and progressive organ dysfunction.6 In the United States, the burden of sickle cell disease has been estimated to cost the health care system more than US$1 billion each year.7
Given the high degree of morbidity and related health care costs associated with a diagnosis of sickle cell disease, an estimated population prevalence with demographic characteristics would be of value in resource allocation planning (i.e., investment in additional comprehensive care programs). Although Simpson and colleagues8 enumerated the number of children in Ontario, Canada, with pathologic hemoglobinopathy, and a more recent effort has been made to register patients with sickle cell disease in the province of Quebec, Canada, who have been diagnosed with COVID-19,9 a comprehensive registry of patients with sickle cell disease in any region of Canada is not yet available. The frequency with which affected patients access acute and emergency care, however, provides an opportunity to estimate the number of unique patients in a defined geographic area.
We therefore performed a study to estimate the number of patients with sickle cell disease in Ontario by querying inpatient and outpatient health administrative databases over a 10-year period. The need for such information has been highlighted by the Sickle Cell Awareness Group of Ontario, a charitable patient organization advocating for patients with sickle cell disease that was engaged in all aspects of this project.
Methods
Setting and data sources
Ontario is Canada’s most populous province, with an ethnically diverse population of about 14 million. It provides access to necessary hospital care and physician services to the large majority of its residents through the publicly funded Ontario Health Insurance Plan (OHIP).
We used 3 health administrative databases to search for patients with a diagnosis of sickle cell disease in Ontario from Apr. 1, 2007, through Mar. 31, 2017: the Discharge Abstract Database (DAD), compiled by the Canadian Institute for Health Information, containing diagnoses and procedures for all admissions to acute care hospitals in Ontario; the National Ambulatory Care Reporting System (NACRS), which includes visits to emergency departments, cancer clinics and dialysis clinics, linked with other data sources to identify transitions to other care settings; and Newborn Screening Ontario, a publicly funded program that screens more than 99% of newborns in Ontario for rare but treatable diseases using a combination of advanced laboratory techniques and that expanded to include sickle cell disease and traits in 2006.10 These data sets were linked using unique encoded identifiers and analyzed at ICES. Each patient record in DAD contains up to 25 diagnoses, and up to 10 diagnoses may be found in NACRS records. We queried these diagnostic fields for the following codes from the Canadian version of the International Statistical Classification of Diseases and Related Health Problems, 10th Revision: D570 (Sickle cell anemia with crisis), D571 (Sickle cell anemia without crisis), D572 (Double heterozygous sickling disorders) and D578 (Other sickle cell disorders). Because the search for these 4 codes was conducted in every diagnostic space within the data sets, all interactions during which sickle cell disease was deemed to have contributed meaningfully to the patient’s treatment were captured. The Newborn Screening Ontario database was queried for newborns diagnosed with hemoglobin SS, S/β-thal, SC, SE or S/HPFH genotypes.
We identified repeat presentations by the same patient within each of the 3 databases by cross-referencing individual OHIP numbers. The OHIP number was also used to link to the Registered Persons Database to obtain basic demographic details, including the postal code on July 1 of each year of query. The postal code was in turn linked to the Postal CodeOM Conversion File Plus (2006, 2011 and 2016) to obtain additional census geographic identifiers such as urban or rural (communities with < 10 000 people) residence, neighbourhood income quintile and regional health zone (i.e., the 14 Ontario Local Health Integration Networks, known as of 2021 as Home and Community Care Support Services corporations).
Statistical analysis
We analyzed the identified cohort of patients descriptively regarding age, sex, location of residence (urban v. rural, regional health zone), and type and number of health care interactions for sickle cell disease during the study period. We calculated the rate of natural increase each year by subtracting the number of deaths from the number of diagnoses in newborns. Health care interactions by those without OHIP coverage (i.e., people who were not Canadian citizens, permanent residents, or landed immigrants or refugees)11 were counted but otherwise were not included in the calculations of disease prevalence owing to lack of a unique identifier by which to distinguish individual patients.
Sensitivity analysis
To determine the robustness of the analysis, we recalculated the cohort size after decreasing the study period from 10 years to 5 years and then to 2 years, and with the sequential exclusion of data from Newborn Screening Ontario and NACRS.
Ethics approval
The use of the data in this project is authorized under section 45 of Ontario’s Personal Health Information Protection Act and did not require review by a research ethics board.12
Results
We identified 44 770 records of patients with a diagnosis of sickle cell disease between Apr. 1, 2007, and Mar. 31, 2017; of these, 1168 (2.6%) did not have an associated OHIP number. Although we could not determine the number of people represented by these non-OHIP visits, their occurrence declined over time (Appendix 1, available at www.cmajopen.ca/content/11/4/E725/suppl/DC1). Non-OHIP interactions were proportionally highest in the Central Toronto and Hamilton regions (8.5% and 7.5% of interactions, respectively) (data not shown).
When we cross-referenced the remaining 43 602 health care interactions by OHIP number, there were 3418 unique patients with a median age of 24 years (interquartile range [IQR] 9–39 yr) at the time of their first captured health care interaction with a diagnosis of sickle cell disease. A slight majority of patients (1912 [55.9%]) were female, although males predominated among those younger than 15 years of age (605 [53.2%]) (Figure 1).

Age distribution of 3418 patients with sickle cell disease identified in Ontario, 2007/08–2016/17.
We identified 492 newborns, with an average rate of natural increase of 35 patients per year (Figure 2). Of the 492 newborns, 295 (60.0%) had Hgb-SS, 130 (26.4%) had Hgb-SC, 53 (10.8%) had Hgb-S/β-thal, 7 (1.4%) had Hgb-SE, and 7 (1.4%) had Hgb-S/HPFH.
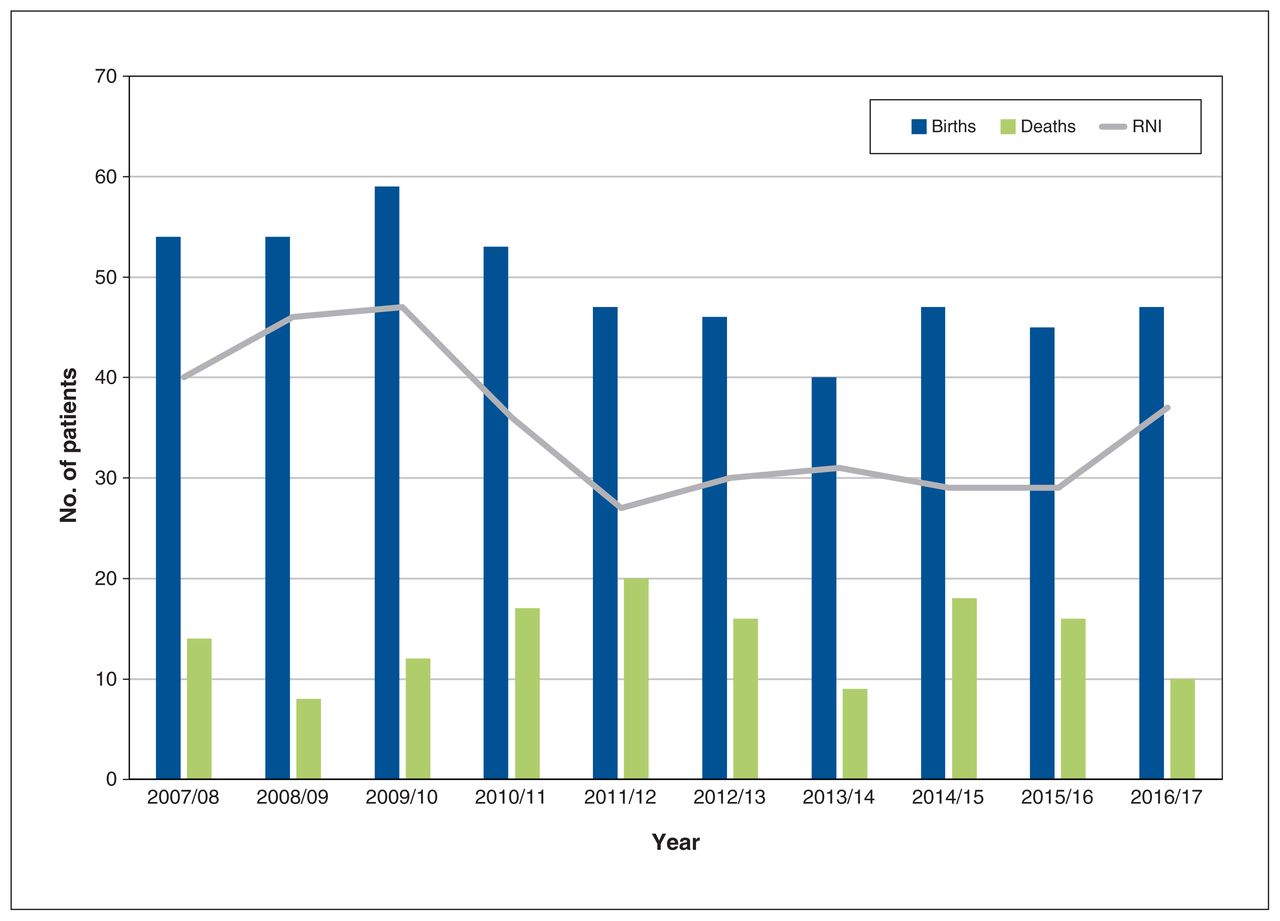
Estimated population change in Ontario patients with sickle cell disease over time. Note: RNI = rate of natural increase, defined as the difference between the number of births and deaths for each year.
Although affected patients were found in every regional health zone in Ontario, they were heavily concentrated in the urban areas of Toronto, Hamilton and Ottawa (Figure 3), and almost double the expected proportion of the cohort lived in neighbourhoods with the lowest income quintile, as compared to the general Canadian population.
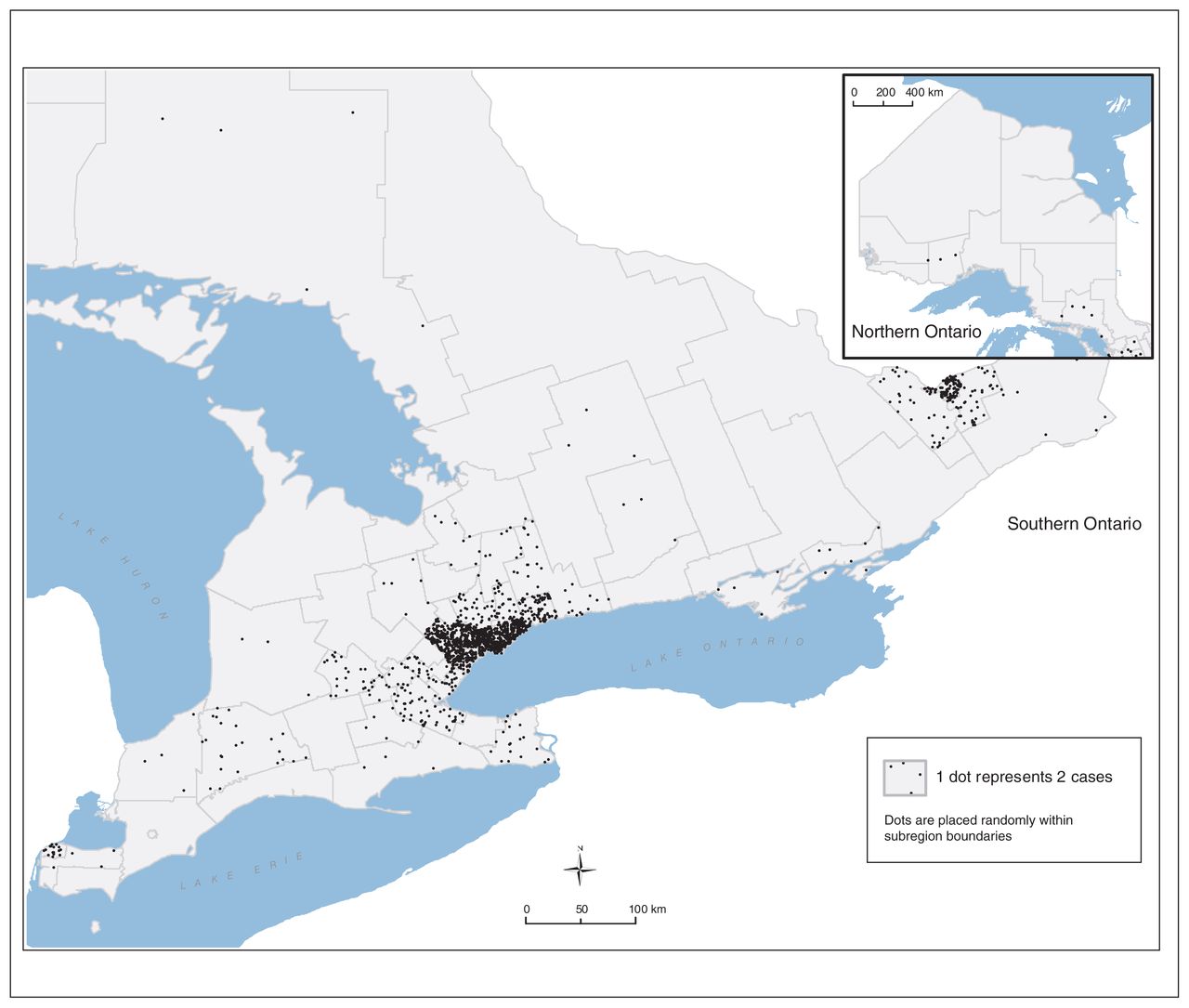
Distribution by geographic area of Ontario patients with sickle cell disease.
A total of 229 patients (6.7%) were documented to have died during the study period, with a mean age at death of 55 years (standard deviation [SD] 21 yr, range 2–93 yr). The average age at the time of death remained stable over the study period.
The pattern of use of health care services was highly variable (Table 1). Over the study period, 14 985 inpatient admissions and 22 800 emergency department visits for sickle cell disease were documented; an additional 4957 non–emergency department outpatient visits (i.e., dialysis and cancer clinic visits) were also documented. The median number of emergency department visits per patient was 2 (IQR 1–7), although the distribution was heavily skewed by a small number of high-use patients, such that the average number of visits was considerably higher (6.69 [SD 26.71]). Similarly, although the median number of hospital admissions per patient over the study period was 1 (IQR 1–5), the average was 4.38 (SD 8.53). A slightly higher proportion of male than female patients were identified by newborn screening (254 [51.6%] v. 238 [48.4%]), and male patients had slightly higher rates than female patients of both emergency department visits and inpatient admissions.
Use of health care services for sickle cell disease among patients with a recorded diagnosis of the disease in Ontario who had any health care contact, 2007/08 to 2016/17
Sensitivity analysis
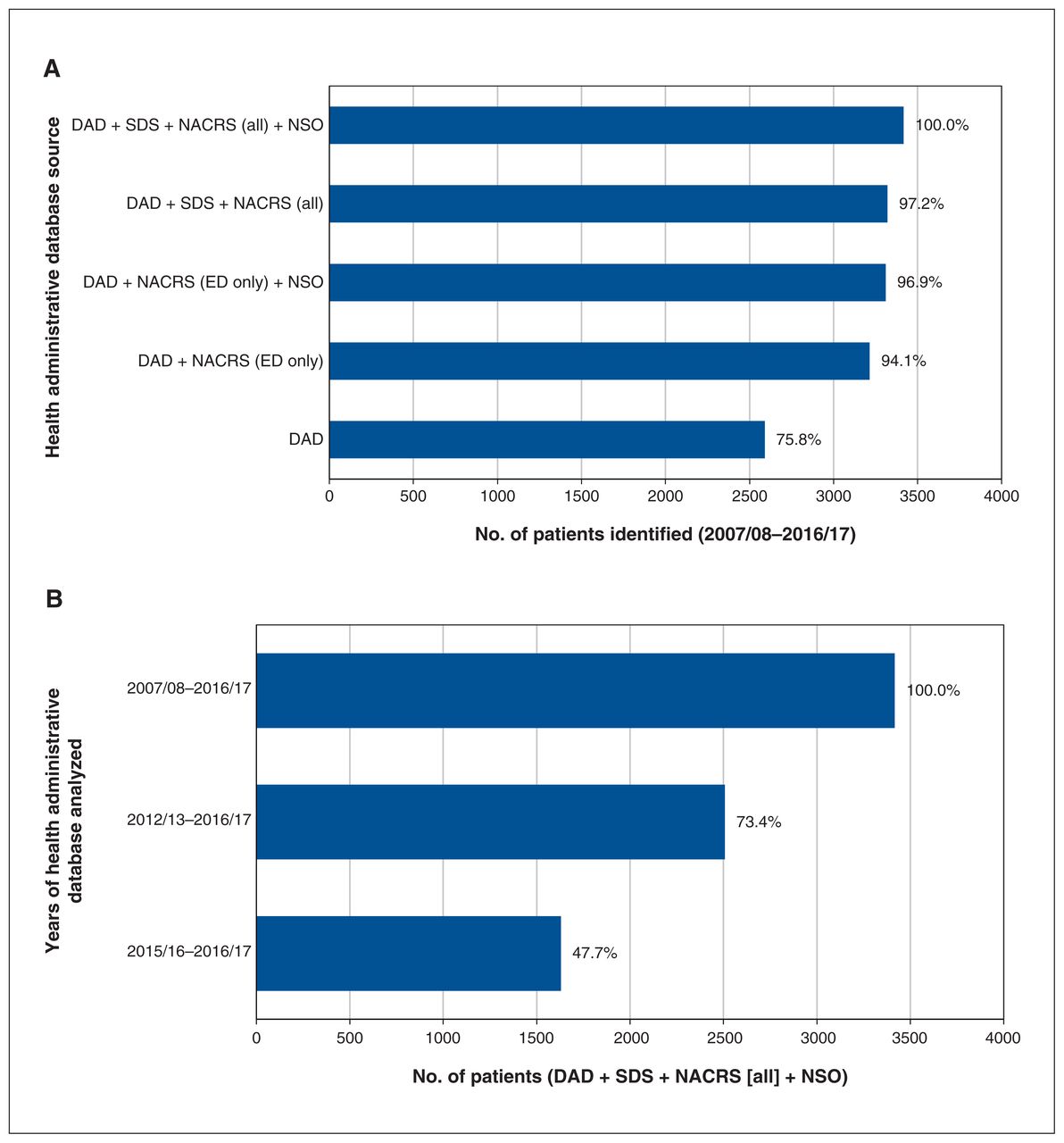
Sensitivity analysis showed that, although excluding non– emergency department outpatient visits from analysis had little effect on the total number of patients captured, limiting analysis to inpatient visits decreased the number of identified patients by nearly one-quarter. There was a similarly large effect when the period of analysis was reduced from 10 years to 5 years, and from 5 years to 2 years (Figure 4). Excluding patients identified by newborn screening resulted in a loss of 95 patients. Although this represented a very small proportion (2.8%) of the cohort, it represented 19.3% of those identified by newborn screening, which indicated that nearly one-fifth of the newborns had no other hospital interactions during the study period.

Sensitivity analysis by (A) health administrative database source and (B) years of health administrative database analyzed. Note: DAD = Discharge Abstract Database, ED = emergency department, NACRS = National Ambulatory Care Reporting System, NSO = Newborn Screening Ontario, SDS = same-day surgery.
Interpretation
The estimated census of 3418 patients with sickle cell disease in Ontario suggests a population prevalence of about 1 in 4200. Although this meets Health Canada’s definition of a rare disease (fewer than 5 in 10 00011), it is notable that sickle cell disease is twice as prevalent as other, better-funded genetic conditions such as hemophilia13 and cystic fibrosis.14 There was a large degree of heterogeneity in the intensity with which patients in this cohort required acute care management during the 10-year study period; however, the absolute numbers of emergency department and inpatient visits related to sickle cell disease were high.
The slight overrepresentation of male patients in birth rates is unexplained, although sex differences in the prevalence of autosomal alleles have also been documented in other conditions.15 Males also appeared to have a worse clinical course. This was evident in the higher rate of emergency department visits and inpatient admissions compared to age-matched female patients, but was also suggested by a transition to a female majority in all cohorts older than 14 years. This finding mirrors that of Baum and colleagues,16 who observed a large increase in vaso-occlusive pain crises among males starting at age 15 years. Other studies have also shown adult male patients with sickle cell disease to have higher rates of pain17 and shorter life expectancy than women with the disease.18 A possible mechanism is a protective effect of estrogens on nitric oxide production, which, in turn, may decrease endothelial dysfunction and increase fetal hemoglobin expression.19
Analysis of health administrative databases is a well-established methodologic technique for approximating the number of patients in a defined geographic area with sickle cell disease.20 For example, Grosse and colleagues20 analyzed Medicaid claims to enumerate the number of pediatric patients with sickle cell disease in studies in Tennessee, Michigan and Georgia. Those studies were validated with the use of data from newborn screening databases and generally showed high sensitivity and specificity, although accuracy varied with the number of interactions required to confirm a case and the length of time the audit was conducted over.
The methodology used in this study could be applied to other provinces in Canada, with some important caveats. First, newborn screening must be in place to identify patients too young to have required inpatient or outpatient medical care for their diagnosis. Although this represented a small proportion of the total population captured in our analysis, it is notable that nearly 1 in 5 patients identified via newborn screening had no other documented interaction with the health care system during the study period. At the time of publication, newborn screening for sickle cell disease was being performed in all regions of Canada except Manitoba and Newfoundland and Labrador, but the number of years of data available for analysis varies; Alberta, the most recent addition, began newborn screening only in 2019.21 A more important consideration is the inclusion of data from out-patient visits. In Canada, this information is captured by NACRS, but the degree to which this database includes visits to emergency departments varies by province.22 In addition, diagnosis and procedure codes are submitted to NACRS with varying degrees of detail. Although inpatient data submitted to DAD are collected more consistently on a national level, our results suggest that relying on this database alone will miss about one-quarter of patients with sickle cell disease when using a 10-year case ascertainment period. Shortening the period of analysis to less than 10 years would result in further loss of sensitivity. The duration of the period required to sustain data capture may increase over time, as the continued provision of comprehensive care to patients with sickle cell disease steadily decreases the frequency of emergency department visits and hospital admissions.23 Future studies may find it more efficient to search for patients with sickle cell disease on the basis of laboratory tests. At the time of the current study, however, the Ontario Laboratories Information System had not been validated for research inquiries of this kind.
We did not directly address the costs of care for patients with sickle cell disease in this study. In an analysis from the US, Ballas24 estimated the lifetime costs for a single patient with sickle cell disease to approach US$9 million. Equivalent costs in the context of the Canadian health care system have yet to be assessed. However, they are likely to be comparable to those estimated in the US and can be expected to increase over time. For example, although the number of affected births in this study appeared to be trending downward over the study period, the rate of natural increase remained positive, at about 35 new cases per year. This means that, even without considering the effects of immigration, the number of affected people is expected to grow. In the present study, the incidence of Hgb-SC disease (generally considered a less severe form of sickle cell disease17) among newborns was about half that observed for Hgb-SS disease, consistent with what has been reported in the US.3 In addition, although the average age at death in our cohort was 55 years — also comparable to what has been estimated in the US25 — the average patient age was 24 years (versus a median of 40 yr for the provincial population26), which suggests that the burden of disease in this population will increase over time as these patients age and accumulate both progressive chronic organ dysfunction and need for transfusion support. Although transfusion data were not captured in the current study, a similar investigation in the US showed a doubling between 2000 and 2013 of hospital stays during which transfusion was the sole procedure,27 and Nouraie and Gordeuk28 reported that one-third of admissions for pain crises in adults in 2007–2012 were accompanied by transfusion. This growing reliance on transfusion support of patients with sickle cell disease may reflect an increasing incidence of disease complications for which transfusion is indicated, but it may also reflect increased awareness of transfusion guidelines.29–31
Limitations
Strengths of our study include capture of all insured patients within a single-payer, public health care system; the inclusion of data from a universal newborn screening program to capture patients too young to have required treatment in an acute care facility; and a 10-year period of surveillance, which allowed the capture of patients who infrequently required acute care. In comparison, attempts to create sickle cell disease registries in other countries have relied on voluntary reporting32,33 or health administrative databases that include only a minority of the population under study.34,35
Our study also has limitations. Neither the diagnostic codes used to gather data nor the search algorithm used were validated against individual patient chart review. However, a study in which a similar methodology was used showed that analysis of Ontario health administrative databases is a reliable method of identifying children with hemoglobinopathy.8 In addition, we did not require multiple interactions to confirm coding accuracy, and, therefore, our findings may be susceptible to false-positive results (i.e., miscoding sickle cell trait as disease).
Limitations intrinsic to the study design may also have resulted in underestimation of the number of patients with sickle cell disease in the province. First, we excluded people without OHIP coverage. Although only a small proportion of health care interactions lacked an associated OHIP number, it is possible that people lacking OHIP coverage are less likely to seek acute care for socioeconomic reasons. Patients who never sought acute care owing to the absence of severe symptoms would also not have been captured, but this is likely a small number. For example, Amendah and colleagues36 found that only 12% of pediatric patients with sickle cell disease never sought treatment over a 5-year follow-up period. Among patients who have health care insurance, there is no evidence that lower socioeconomic status results in underuse of health care services. On the contrary, such patients tend to have greater reliance on acute care support owing to inadequate access to preventive health care and increased exposure to environmental exacerbators of their disease.37,38 Although newborns born at any time during our 10-year surveillance period would have been captured in our analysis, patients who immigrated to Ontario after 2007 effectively received less than 10 years of surveillance for acute care interactions and therefore were more likely to have been missed. Finally, the consistently positive rate of natural increase observed suggests that the population of patients with sickle cell disease in Ontario has likely increased since 2016/17, the final year of the surveillance period.
Conclusion
In 2007/08 to 2016/17, there were about 3500 people with sickle cell disease in Ontario, and the number increased over that period. Although most patients lived in urban areas, they resided in every region of the province and had substantial, although highly variable, reliance on hospital-based care. In the absence of a formal patient registry or validated provincial laboratory information system, the methodology we adopted likely represents the most feasible means of monitoring the prevalence of this disease in Ontario and other Canadian provinces. Estimates of sickle cell disease prevalence and burden can be used by advocacy groups such as the Sickle Cell Awareness Group of Ontario and government agencies to inform funding decisions, best-care practices and policy development.
Footnotes
Competing interests: None declared.
This article has been peer reviewed.
Contributors: All of the authors contributed to the conception and design of the study. Eliane Kim analyzed the data. Jacob Pendergrast wrote the manuscript. All of the authors contributed to data interpretation, revised the manuscript critically for important intellectual content, approved the final version to be published and agreed to be accountable for all aspects of the work.
Funding: With the support of the Ontario Ministry of Health and Ministry of Long-Term Care, this project was funded by the ICES Applied Health Research Question Program.
Data sharing: The data set from this study is held securely in coded form at ICES. Although legal data-sharing agreements between ICES and data providers (e.g., health care organizations and government) prohibit ICES from making the data set publicly available, access may be granted to those who meet prespecified criteria for confidential access, available at https://www.ices.on.ca/DAS (email: das{at}ices.on.ca). The full data set creation plan and underlying analytic code are available from the authors on request, with the understanding that the computer programs may rely on coding templates or macros that are unique to ICES and are therefore either inaccessible or may require modification.
Supplemental information: For reviewer comments and the original submission of this manuscript, please see www.cmajopen.ca/content/11/4/E725/suppl/DC1.
Disclaimer: Parts of this material are based on data and/or information compiled and provided by the Canadian Institute for Health Information (CIHI) and the Ontario Ministry of Health. Geographic variables were adapted from the following Statistics Canada products: Postal CodeOM Conversion File Plus (PCCF+) version 5E (2006), PCCF+ version 6D (2011) and PCCF+ version 7B (2016). This does not constitute an endorsement by Statistics Canada of this material. The analyses, conclusions, opinions and statements expressed herein are solely those of the authors and do not reflect those of the funding or data sources; no endorsement is intended or should be inferred. No funding bodies had any role in the study design, data collection, analysis, decision to publish or preparation of the manuscript.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
- © 2023 CMA Impact Inc. or its licensors
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools





Related Articles
Cited By...
- No citing articles found.