


Abstract
Background: Genetic testing in families with hereditary cancer enables identification of people most likely to benefit from intensive screening and preventive measures; however, the uptake of testing in relatives (known as cascade carrier testing) for hereditary colorectal cancer syndromes has been shown to be low. Our objective was to report rates of familial testing for hereditary colorectal cancer syndromes in a publicly funded hereditary cancer clinic in Canada.
Methods: A cross-sectional retrospective database review was used to determine testing uptake between 1997 and 2016 for families served by the provincial Hereditary Cancer Program for British Columbia and Yukon. Analyses were conducted for genes associated with syndromes with an increased risk for colorectal cancer, including Lynch syndrome (MLH1, MSH2, MSH6, PMS2 and EPCAM) and familial adenomatous polyposis (APC), and for additional moderate- to high-penetrance genes (STK11, TP53, SMAD4, MUTYH, PTEN and CHEK2). Descriptive statistics were used and all analyses were 2-tailed.
Results: The study cohort included 245 index patients, with carrier testing performed in 382 relatives. The mean age at family member testing was 41.2 years, and most (61.0%) of the family members who underwent testing were women. The median time between disclosure of index cases and their family member’s results was 8.3 months. Among eligible first-degree relatives, 32.6% (268/821) underwent testing in BC. Of 67 cancer diagnoses in family members, most (62.7%) occurred before genetic testing.
Interpretation: A substantial proportion of people at risk for hereditary colorectal cancer do not undergo genetic testing. This gap highlights the need to explore barriers to testing and to consider interventions to promote uptake; more aggressive efforts by hereditary cancer programs are needed to reach this highest risk population.
It is estimated that 5%–6% of colorectal cancers are due to a hereditary cancer syndrome.1,2 Genetic testing in at-risk relatives, known as cascade carrier testing, identifies those most likely to benefit from increased screening and preventive measures, leading to earlier detection and decreased cancer incidence.3 The cost-effectiveness of broad panel genetic testing relies on uptake of targeted carrier testing and arguably on the effectiveness of hereditary cancer programs in general.4–6
The most common hereditary colorectal cancer syndrome, Lynch syndrome, accounts for up to 5% of colorectal cancers.2 People with Lynch syndrome have up to an 80% and 60% lifetime risk (up to age 80 yr) for colorectal and endometrial cancer, respectively.7 In people with more rare hereditary colorectal cancer syndromes such as familial adenomatous polyposis and MUTYH-associated polyposis, there is up to a 100% colorectal cancer lifetime risk (to age 80 yr) without appropriate surveillance.8 Given the substantially higher cancer risk among people with these syndromes than in the general population, identifying those at risk is an important health care priority.9
Reports suggest variable carrier testing rates in clinic-based and cancer registry–based studies of at-risk relatives, ranging from 34% to 75%.4,10–23 Most people who have received carrier testing for Lynch syndrome are satisfied in the long term with receiving testing,11 as carrier testing enables them to make more informed decisions and to receive personalized health care.24 Noncarriers also benefit from carrier testing, as negative test results may reduce cancer-related anxiety25 and can eliminate the need for intensive screening.16 Many studies have found that substantially higher numbers of women than men receive carrier testing.12,16,21 Some studies have also related age to carrier testing uptake, with lower rates of testing in first-degree relatives younger than 25 years of age for Lynch syndrome21 and older than 40 years of age for familial adenomatous polyposis.16
To understand the uptake of carrier testing in a Canadian context, our objective was to report rates of familial testing for hereditary cancer syndromes associated with an increased risk for colorectal cancer in a publicly funded hereditary cancer clinic over a 20-year period.
Methods
Setting and study population
This study was a retrospective cross-sectional database review using population-based data from the Hereditary Cancer Program (HCP) for British Columbia and Yukon. This program is the sole provider of publicly funded cancer genetic testing in these jurisdictions. Demographic data and personal medical and family history information for patients assessed between Jan. 1, 1997, and Dec. 31, 2016, were obtained using the HCP clinical and BC Cancer electronic chart databases.
Individuals found to carry a pathogenic or likely pathogenic variant (i.e., the index patient) tested through the HCP program were included in the study. Their family members known or presumed to be living in BC or Yukon were included in the cascade carrier analysis. Family members confirmed to be living outside of British Columbia or Yukon were excluded from the analysis, although it should be noted that geographic location of unaffected family members is not routinely assessed or recorded in the HCP database.
The index patients and their family members were identified through the HCP clinical database; this database is maintained using the software Progeny, a genetic pedigree and medical record program commonly used in medical genetics. Previous research experience with these data sets has indicated high levels of consistency between clinical and genetic diagnoses in Progeny and the BC Cancer database (Matthew Richardson, Hereditary Cancer Program, BC Cancer, Vancouver, BC: personal communication, 2020).
The genetic variants assessed were all linked to a risk for hereditary colorectal cancer. The autosomal dominant conditions Lynch syndrome (MLH1, MSH2, MSH6, PMS2 and EPCAM), familial adenomatous polyposis (APC), Li-Fraumeni syndrome (TP53), Peutz-Jeghers syndrome (STK11), juvenile polyposis (SMAD4) and PTEN hamartoma tumour syndrome (which includes Cowden syndrome; PTEN), and CHEK2 where first-degree relatives of gene-positive individuals have a 50% risk of carrying the familial pathogenic variant, were included.16
Additionally, we included the autosomal recessive syndrome MUTYH-associated polyposis, where at-risk relatives may have a 25% risk of homozygous or biallelic MUTYH pathogenic variant status.16 For this syndrome, families identified through index cases with biallelic MUTYH pathogenic or likely pathogenic variants were included in the analysis, with only siblings deemed as eligible individuals for testing.
For adult-onset syndromes (Lynch, Peutz-Jeghers, juvenile polyposis, PTEN hamartoma tumour syndrome [which includes Cowden syndrome] and CHEK2), relatives were considered eligible for carrier testing if they were 19 years of age or older and living in BC or Yukon. In the case of childhood-onset syndromes (Li-Fraumeni, familial adenomatous polyposis), living relatives of all ages were eligible.
Cascade carrier testing
In-depth pedigree analyses were performed for each index patient to ascertain the number of eligible first-degree relatives and the number of relatives tested by degree (first through fourth degree). The pedigrees and test results were recorded as part of routine medical care in the HCP database, while the determination of eligible relatives versus actual relatives tested was performed for the study (V.B.). Relatives’ eligibility for testing was determined by existing HCP clinical criteria. In the case of multiple tests performed in 1 family, the first individual tested was considered the index patient. The median time between the index and carrier tests per gene was determined by calculating the difference between the date of disclosure of index and all carrier test results.
Cohort characteristics
Summary statistics were calculated to describe the population of index patients and relatives receiving carrier tests by age, sex, rural or urban residence and referral method. The location of the patient’s residence (urban v. rural) was determined from their 6-digit postal code. The types and number of cancers diagnosed in patients before and after carrier testing were extracted from the BC Cancer database and assessed to determine the health impact of carrier testing. We included all cancer types in this analysis, including nonmelanomatous skin cancers and cervical cancer.
Statistical analysis
Descriptive statistics were calculated using Microsoft Excel 2016. Results were reported as means and standard deviations or medians and ranges for continuous variables and as proportions or frequencies for categorical variables. R version 3.3.4 was used to perform χ2 tests to determine uptake by sex and age as well as to perform univariate and multivariate analyses to assess the relationship of age, sex, urban or rural residence and cancer diagnosis with testing uptake. Univariate and multivariate analyses were employed to assess factors related to the time interval between index and carrier tests. All statistical analyses were 2-tailed with a statistical significance of p less than or equal to 0.05.
Ethics approval
This study was reviewed and approved by the University of British Columbia – BC Cancer Research Ethics Board and the Human Research Ethics Board of the University of the Fraser Valley.
Results
The study cohort included 245 index patients tested between Jan. 1, 1997, and Dec. 31, 2016. During this period, carrier testing was performed for 382 relatives of the index patients (first through fourth degree). Demographic data and source of referral for the index patients and relatives receiving carrier testing are reported in Table 1.
Demographic characteristics of people who received index or carrier testing in British Columbia or Yukon through the BC Hereditary Cancer Program, 1997–2006
For the index patients, the cohort included 150 (61.2%) women and 95 (38.8%) men. Pathogenic or likely pathogenic variants were identified in Lynch syndrome genes for 157 (64.1%) of the index patients. The mean age at index testing was 49.3 (standard deviation [SD] 15.1) years, and 209 (85.3%) and 24 (9.8%) patients lived in urban and rural areas, respectively (Table 1).
Among the 382 family members who underwent carrier testing, 233 (61.0%) were women and 149 (39.0%) were men. The mean age at carrier testing was 41.2 (SD 17.7) years. Among those who underwent carrier testing, 332 (86.9%) lived in urban regions; 35 (9.2%) lived in rural regions.
The most common source of referral was a medical specialist for the index patients (129 patients, 52.7%) and self-referral for those undergoing carrier testing (185 family members, 48.4%). Self-referral was the only characteristic of index patients that was significantly correlated with increased carrier testing in first-degree relatives (p = 0.03).
Cascade carrier testing
Figure 1 shows the uptake of carrier testing in eligible first-degree relatives of index patients in BC and Yukon. Of 821 first-degree relatives eligible for testing, 268 (32.6%) received carrier testing. The highest uptake was half of all eligible first-degree relatives for CHEK2 (5/10; 50.0%, 95% confidence interval [CI] 19% to 81%) and SMAD4 (3/6; 50.0%, 95% CI 10% to 90%), and the lowest uptake was 10.7% of first-degree relatives of MUTYH-associated polyposis (3/28, 95% CI −1% to 22%). We identified 91 families with eligible first-degree relatives in BC and Yukon with no carrier tests performed.
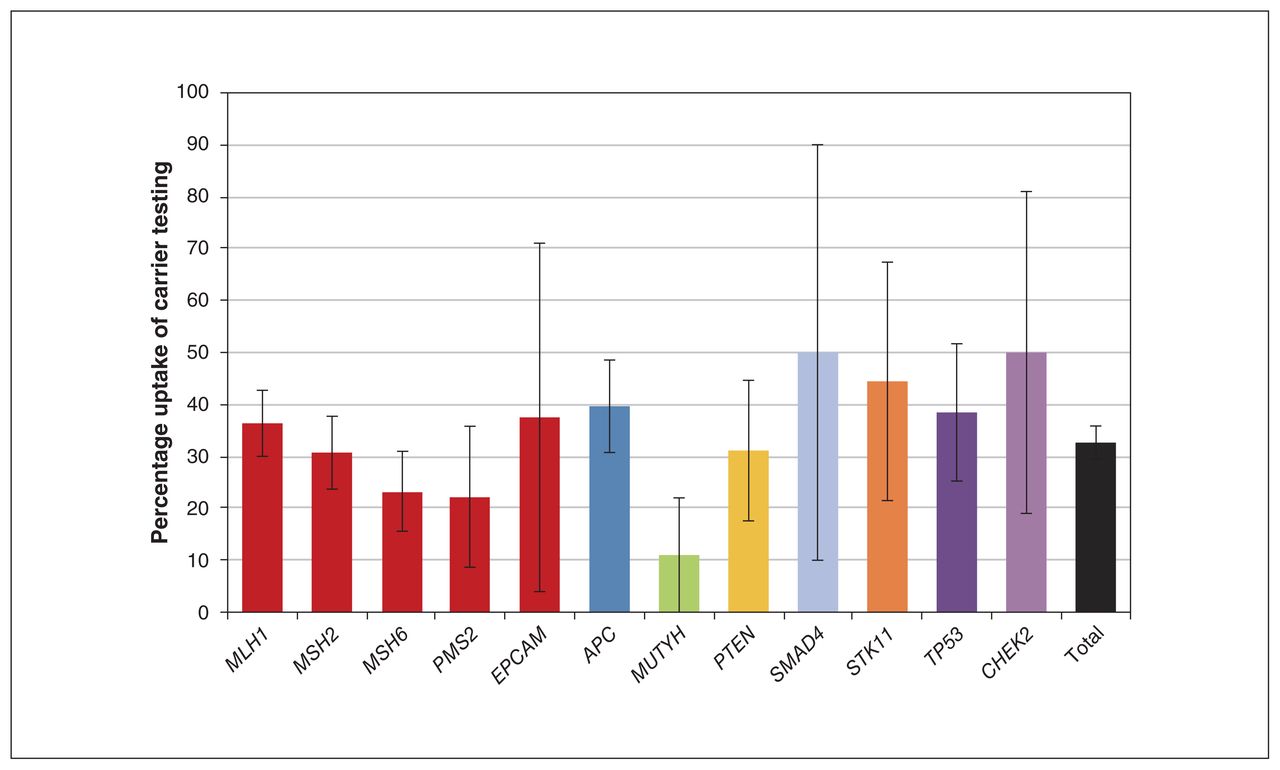
Percentage of eligible first-degree relatives who have received carrier testing. See Appendix 1 (available at www.cmajopen.ca/content/8/4/E637/suppl/DC1) for raw data. Of 821 eligible first-degree relatives, 268 received carrier testing. Error bars represent 95% confidence intervals.
The total cascade carrier rate across all genes among all relatives receiving carrier testing was 1.56 carrier tests per index patient (382/245) (Table 2). The median time to carrier testing was 8.3 (range 0–170.3) months. The median time to testing for the 5 Lynch syndrome genes was 8.2 (range 0–143.0) months. The age of the family member was significantly correlated with the time to uptake of carrier testing, with younger family members having a longer time gap before pursuing testing (p < 0.001). Among relatives who were women and those who referred themselves to the program, a trend toward shorter time to uptake for testing was observed; however, these findings were not statistically significant.
Characteristics of recipients of carrier testing (first- to fourth-degree relatives) and time difference between index and carrier testing, by gene tested
Cancer diagnoses
A total of 67 cancer diagnoses were identified in carriers of a colorectal cancer–related gene. Colorectal cancer was the most commonly diagnosed type of cancer (n = 32; 47.8%), followed by breast cancer (n = 8; 11.9%) and endometrial cancer (n = 7; 10.4%). Most cancer diagnoses (n = 42; 62.7%) occurred before carrier testing; 25 of the diagnoses (37.3%) occurred after testing.
Interpretation
In this retrospective analysis, we assessed the uptake of genetic testing in 245 families with a known familial pathogenic or likely pathogenic variant in a colorectal cancer–related gene. The index patients from these families were ascertained from our general clinic population, which has an approximately 11% mutation detection rate across these genes (Matthew Richardson, Hereditary Cancer Program, BC Cancer, Vancouver, BC: unpublished data, 2020). Across the 12 genes analyzed, the uptake of testing was 1 in 3 eligible first-degree relatives. The most common genes tested for were those related to the relatively common Lynch syndrome, which comprised 64.1% of index diagnoses; overall uptake was highest in female relatives. The uptake of testing for Lynch syndrome in first-degree relatives in previous reports has ranged between 34% and 94%.14–16,20–22
To date, reports on cascade carrier rates have differed widely across studies. We identified a rate of 1.56 carrier tests per index case across all 12 genes. Similar rates of 1.514 and 1.0413 relatives tested per index patient have previously been reported for Lynch syndrome. However, other studies have reported rates of 4.626 and 3.616 carrier tests per index patient, demonstrating large variability in cascade carrier testing between populations. This variability highlights the value of a population-based approach to assessing carrier testing, as uptake rates may differ greatly between clinics and countries.
The proportion of patients from rural areas in this study was lower than we might have expected on the basis of census data. During the study period (1997–2016), 9.2% of family members completing carrier testing for hereditary colorectal cancer were from rural regions in BC; this percentage is lower than the overall percentage of BC residents who lived in rural areas in 1996 (18%)27 and 2016 (14%).28 Some studies have shown disparities in awareness among rural populations,29 whereas others have demonstrated higher uptake of testing when alternative methods like telephone counselling are used.30 These findings support the development and use of alternative modes for education service delivery to rural populations to ensure equitable access. They also highlight the importance of patient self-referrals. Many programs across Canada require a physician referral for hereditary cancer services. Canadians living in rural areas have decreased access to family physicians31 and thus face even greater barriers to receiving specialty care if physician referral is required. Our study showed greater numbers of tests in families that started with our program through self-referral and a trend toward a shorter wait time when they initiated their own referral (although this latter finding was not statistically significant).
A previous cancer diagnosis has been identified as a predictor of carrier testing.32 We found that nearly two-thirds (62.7%) of cancer diagnoses in carriers were diagnosed before carrier testing, similar to the percentage found in another Canadian study on Lynch syndrome (63.4%).33 Considering that a major goal of carrier testing is cancer prevention through intensive screening and prophylactic measures, it is important to reach at-risk relatives before they are diagnosed with cancer.34 Further investigation into the health impact of carrier testing for colorectal cancer–related syndromes is necessary.
Fears about life insurance and mortgage implications are commonly reported reasons for declining carrier testing.18 In 2017, Canada enacted the Genetic Non-Discrimination Act, preventing insurance companies, employers and people involved in any contract from requiring individuals to disclose genetic test results.35 The law was upheld in 2020 by the Supreme Court of Canada after a challenge related to jurisdictional concerns.36 Research investigating carrier testing rates before and after enactment will be an important subject of future research.
Recent systematic reviews on cascade carrier screening point to the limitation of the index patient being responsible to inform all at-risk relatives.9,23 Although this remains the standard procedure in most hereditary cancer programs, including that of BC Cancer, studies where health professionals also contacted relatives have reported higher rates of testing uptake.15,16,20–22 Policy and legal considerations are needed when proposing direct contact with relatives, making this an area for further research.
Our results highlight the need to explore barriers and develop tailored interventions to promote testing uptake. Further research is also needed to examine the effect of demographic factors such as socioeconomic status and ethnicity on uptake of testing, as there is evidence of disparities in cancer screening among Canadians of minority and lower socioeconomic backgrounds.38 This will help guide decision-making regarding education and resource allocation to optimize carrier testing uptake.
Limitations
An important limitation of this study is that we included only index tests and associated carrier tests conducted in BC and Yukon in the analyses to ensure accuracy of data. However, many carrier tests have been performed through the HCP for family members for whom the initial family member who tested positive resided outside of BC or Yukon, and these families were not included in our study. We are likely to have overestimated the number of family members living in BC or Yukon. The geographic location of unaffected relatives is not routinely assessed or recorded in the HCP health record. Therefore, it is possible that some at-risk relatives received out-of-province carrier testing for which we did not have records. The cascade carrier testing rate may be higher than determined, because we were not able to precisely determine the percentage of first-degree relatives living in BC or Yukon.
Additionally, cancer diagnoses included only those for carriers living in BC or Yukon because of limited access to data for cancer diagnoses for family members living elsewhere. The diagnoses we included in the study included all types of cancer (e.g., nonmelanomatous skin cancers and cervical cancer), not just those specific to the colorectal syndromes.
Although our data captured index and carrier tests performed over 20 years, people tested between 2017 and the time of data collection were not included in the analyses. Future analyses that assess carrier testing after 2016 will serve as valuable contributions to the growing body of carrier testing literature.
Although this study was performed in a single program in 1 province and territory, our estimation of cascade testing rates is probably still helpful. British Columba is the largest province in Canada with a single hereditary cancer program serving all residents; such an analysis in other large provinces may be difficult given the fragmentation of hereditary cancer care across several regional programs.
Conclusion
In this study, we found that a substantial proportion of people at risk for hereditary colorectal cancer in BC and Yukon have not received carrier testing. Studies that explore barriers to testing in this population may elucidate avenues for interventions to promote testing, with the ultimate goals of early detection and prevention of hereditary cancer.
Acknowledgment
The authors thank the patients and families of the Hereditary Cancer Program of BC Cancer.
Footnotes
Competing interests: None declared.
This article has been peer reviewed.
Contributors: Vivienne Beard, Angela Bedard, Jennifer Nuk, James Bedard, Sophie Sun and Kasmintan Schrader conceived the study. Vivienne Beard, Angela Bedard, Petra Lee and Quan Hong curated and analyzed the data. Vivienne Beard and Angela Bedard wrote the original draft of the article, which all authors revised. All authors approved the final version to be published and agreed to be accountable for all aspects of the work. Vivienne Beard and Angela Bedard are joint first authors. Sophie Sun and Kasmintan Schrader are joint senior authors.
Funding: Kasmintan Schrader is supported by the Michael Smith Foundation for Health Research and the Canadian Institutes of Health Research.
Data sharing: The data that support the findings of this study are available in aggregate form on request from the corresponding author. The data are not publicly available because of privacy or ethical restrictions.
Supplemental information: For reviewer comments and the original submission of this manuscript, please see www.cmajopen.ca/content/8/4/E637/suppl/DC1.
References
- Copyright 2020, Joule Inc. or its licensors
In this issue

{kind=link}
Article tools





Related Articles
Cited By...
- No citing articles found.