Article Text

Abstract
Background The contribution of copy-number variation (CNV) to disease has been highlighted with the widespread adoption of array-based comparative genomic hybridisation (aCGH) and microarray technology. Contiguous gene deletions involving ANKRD11 in 16q24.3 are associated with autism spectrum disorder (ASD) and intellectual disability (ID), while 16q24.1 deletions affecting FOXF1 are associated with congenital renal malformations, alveolar capillary dysplasia, and various other abnormalities. The disease associations of deletions in the intervening region, 16q24.2, have only been defined to a limited extent.
Aim To determine whether deletions affecting 16q24.2 are correlated with congenital anomalies.
Methods 35 individuals, each having a deletion in 16q24.2, were characterised clinically and by aCGH and/or SNP-genotyping microarray.
Results Several of the 35 16q24.2 deletions identified here closely abut or overlap the coding regions of FOXF1 and ANKRD11, two genes that have been previously associated with the disease. 25 patients were reported to have ASD/ID, and three were found to have bilateral hydronephrosis. 14 of the deletions associated with ASD/ID overlap the coding regions of FBXO31 and MAP1LC3B. These same genes and two others, C16orf95 and ZCCHC14, are also included in the area of minimal overlap of the three deletions associated with hydronephrosis.
Conclusions Our data highlight 16q24.2 as a region of interest for ASD, ID and congenital renal malformations. These conditions are associated, albeit without complete penetrance, with deletions affecting C16orf95, ZCCHC14, MAP1LC3B and FBXO31. The function of each gene in development and disease warrants further investigation.
- Hydronephrosis
- Copy-number
- FBXO31
- MAP1LC3B
Statistics from Altmetric.com
Introduction
Contiguous gene deletions at 16q24 are associated with disease. In 16q24.3, deletions affecting the coding regions of three genes, GALNS, APRT and ANKRD11, have been identified in individuals with congenital disease/anomalies (figure 1). Wang et al (1999)1 described a 100-Kb deletion affecting the coding regions of GALNS and APRT in an individual with 2,8-dihydroxyadenine urolithiasis Online Mendelian Inheritance in Man (OMIM) #102600) and Morquio disease A (OMIM #253000). Patients with contiguous gene deletions affecting ANKRD11 exhibit a constellation of symptoms consistent with KBG syndrome (OMIM #148050), including intellectual disability, dysmorphic facial features and skeletal abnormalities.2–7 Haploinsufficiency of ANKRD11 is likely the primary aetiology of the congenital abnormalities in these individuals as Sirmaci et al (2011) showed that nonsense mutations within ANKRD11 are associated with KBG syndrome.

16q24 deletions reported in this study. A schematic representation of 16q24 (chr16 : 82 808 200–88 879 649; hg18), showing RefSeq genes within the region, previously reported cases of 16q24 deletions (A), the 35 deletions identified in this study (B) and deletions found on the DECIPHER database (C). Deletions are colour-coded according to the associated phenotype (see Legend). The grey column delineates 16q24.2, the region of interest in this report. The blue column represents the extent of the coding region of ANKRD11. The magenta column marks the coding region of FOXF1. Only the six deletions identified by Stankiewicz et al (2009)8 that were associated with congenital renal malformation are shown.
Deletions in 16q24.1 have also been associated with disease. Stankiewicz et al8 described a series of 10 individuals who harboured deletions, ranging in size from 131 Kb to 3.5 Mb, in this region (figure 1). Each of these individuals was diagnosed with alveolar capillary dysplasia, a rare and lethal disease affecting the microanatomy of the lung. A subset of these individuals was also found to have renal anomalies, including bilateral pelviectasis and hydronephrosis. The authors ascribed these anomalies to haploinsufficiency of FOXF1, a gene mapped to 16q24.1. The coding region of this gene is included in the area of minimal overlap of eight of the 10 deletions that they identified. Furthermore, they showed that heterozygous mutations in FOXF1 are associated with alveolar capillary dysplasia and renal anomalies.8
It is unclear whether deletions affecting the intervening region, 16q24.2, are associated with disease. At present, only a single case report has made such an association.9 In the report, Butler et al9 identified a 265-Kb deletion (figure 1) in an individual with intellectual impairment, microcephaly, distichiasis, bilateral vesicoureteral reflux and glomuvenous malformations. The deletion overlapped the coding region of four genes: C16orf95, FBXO31, MAP1LC3B and ZCCHC14. The authors reasoned that haploinsufficiency of any one or combination of the four genes may contribute to the individual's renal pathology. Further work is needed to validate the pathogenicity of deletions in 16q24.2 and to identify the candidate genes associated with disease.
Here we present a series of 35 individuals each of whom had a deletion in 16q24.2. In all, 25 of these individuals were reported to have some form of neurodevelopmental disorder. A total of 14 of the deletions in these individuals share an interval of minimal overlap that corresponds with the coding regions of FBXO31 and MAP1LC3B, specifically implicating these genes in neurodevelopmental disorder. We further suggest that these two genes, as well as C16orf95 and ZCCHC14, may be associated with congenital renal malformation as they are deleted in three of four patients for whom appropriate diagnostic imaging is available. Collectively, our data implicate contiguous gene deletions in 16q24.2 in neurodevelopmental disorder and congenital renal malformation.
Methods
Study subjects
A total of 35 individuals (17 male, 18 female), each bearing a deletion at 16q24.2, were included in this study. Patient 1 was recruited at the University of Alberta, Edmonton, for the indication of congenital renal malformation. Patients 2–35 were screened at three different clinical genetic diagnostic centres. Patients 2 and 3 were identified at the Hospital for Sick Children, Toronto. Patients 4–6 and 14–35 were identified at Signature Genomic Laboratories, and Patients 7–13 were identified at the Mayo Clinic. The patients referred for microarray testing at these centres demonstrated a neurodevelopmental disorder and/or multiple congenital anomalies. Phenotypic data for each patient were obtained from the referring physician or from the microarray request form. Patients 16 and 17 are siblings.
Control samples
To assess the prevalence of copy-number variations (CNVs) at the 16q24.2 locus in the general population, we inspected microarray data from 11 019 population-based, control individuals, analysed as previously described.10–12 This data set included 4783 controls from the Wellcome Trust Case Control Consortium,13 1123 German controls,14 1234 controls from Ottawa,15 1056 individuals from the HapMap project,16 1287 controls from the Study of Addiction: Genetics and Environment consortium,17 1120 population controls from Ontario18 and 416 individuals from the Ontario Population Genomics Platform.19
Genotype analysis
Genomic DNA was extracted from peripheral blood drawn from each patient. Samples were analysed by array-based comparative genomic hybridisation (aCGH) or high-density SNP array methods. Patient 1's sample was analysed using the Illumina Omni 2.5 M–quad BeadChip microarray platform. Genomic DNA from Patients 4–6 and 15–35 was analysed by oligonucleotide-based aCGH using a 105 K-feature platform (SignatureChipOS V.1.0) or one of two versions of a 135K-feature platform (SignatureChipOS V.2.0 and V.3.0) as previously described.20, 21 Genomic DNA from Patient 14 was analysed by bacterial artificial chromosome-based aCGH using an 1887-clone targeted array (SignatureChip V.4.0).22 Samples from Patients 2 and 3 were analysed using the 4×180 K ISCA V.2 microarray (Agilent). Patients 7–13 were analysed using either the Agilent ISCA 44 K or 180 K microarrays.
Results
Deletions identified in 16q24.2
We report 35 individuals who each harbour a deletion in 16q24.2 (table 1, figure 1). Eight of the 35 patients bear additional, potentially pathogenic CNVs outside of 16q24 (see online supplementary table S1).
Summary of the available clinical information for the 35 patients presented in this report as well as information on the 16q24 deletion identified in each individual
The 16q24 deletions range in size from 27 189 to 2 645 900 bp (table 1). In all, 26 of the deletions are restricted to 16q24.2 (85 600 001–87 200 000, hg18; see online supplementary table S2 for hg19 coordinates). Patient 1's deletion, which is the largest identified in this study, extends from 16q24.1 to 16q24.3. We identified 21 deletions in control DNA samples that overlap Patient 1's deletion (see online supplementary table S3). Notably, none of these deletions are exonic for the 16q24.2 genes FBXO31, MAP1LC3B, ZCCHC14 and C16orf95.
The deletions in Patients 1, 22, 30 and 31 overlap the gene ANKRD11 in 16q24.3 (blue column in figure 1). The coding region of FOXF1, a disease-associated gene in 16q24.1,8 is completely deleted in Patient 6 (magenta column in figure 1). Both of these loci are unaffected in the other patients in this study.
By querying the DECIPHER database (V.5.1, https://decipher.sanger.ac.uk/; table 1), we identified 16q24.2 deletions in three other individuals (Patients 255327, 255929 and 251801) who are not included among the 35 patients referred to above. Each of these deletions extends into 16q24.3 and overlaps the coding region of ANKRD11 (figure 1).
Parental samples were available for inheritance testing for 12 of the 35 deletions reported in this study (table 1). The deletions identified in six patients (1, 3, 8, 10, 21 and 25) all occurred de novo. Maternal inheritance of the deletions was identified in three patients (16, 17 and 22), while for Patient 2, the mother was mosaic (48/200 nuclei) for the deletion. The deletions in two patients (12 and 29) were inherited paternally. All three CNVs found in the DECIPHER database were de novo.
Clinical features of study subjects
Phenotypic data for all 35 patients are summarised in table 1. Detailed clinical reports were obtained for Patients 1 and 2 (see online supplementary information), whereas phenotypic data for Patients 3–35 were limited to the indications for genetic testing outlined in the microarray request form. Photographs of Patient 1 are presented in online supplementary figure S1.
Data on neurocognitive status were available for 27 patients. Of these, 25 patients demonstrated some form of disorder (table 1). Three patients had intellectual disability, which is defined as a subaverage IQ (<70) by the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV).23 A total of 22 patients had autism spectrum disorder (ASD), which we further subdivide into pervasive developmental delay (19/22) and autism (3/22) as per the DSM-IV.23 In all, 11 patients demonstrated craniofacial dysmorphia. The remaining 16 patients either did not demonstrate craniofacial abnormalities (1/16) or relevant phenotypic data were unavailable (15/16). The high prevalence of neurodevelopmental disorder and craniofacial dysmorphia among the study cohort likely reflects ascertainment bias. Both of these conditions are common indications for microarray testing at the various genetic diagnostic centres at which patients in this study were identified.
Deletions associated with neurodevelopmental disorders and congenital renal malformation
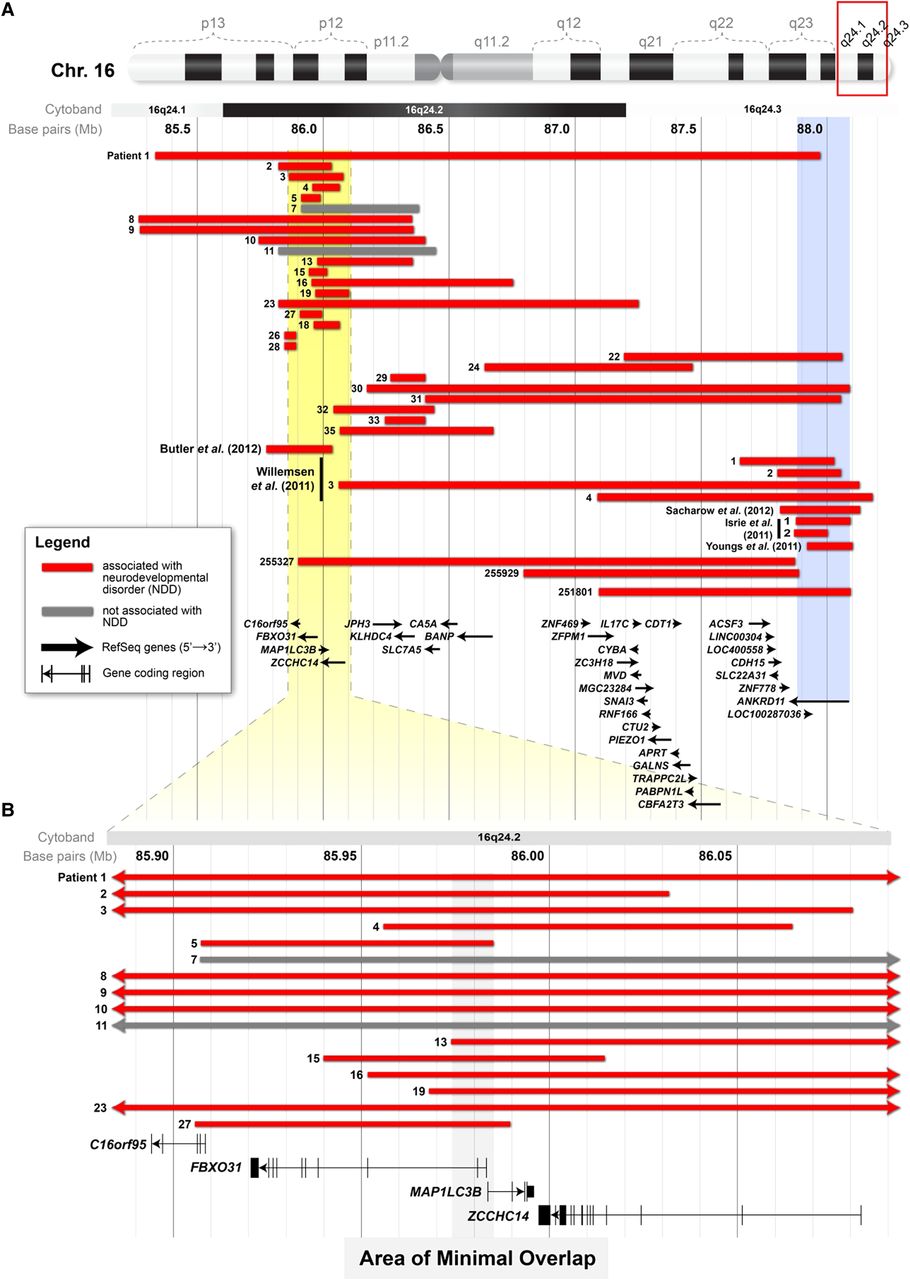
The deletions in individuals with neurodevelopmental disorders are heterogeneous in terms of size and their location within 16q24.2 (figure 2). The deletion in Patient 1 spans the entire length of 16q24.2 and overlaps the other 24 deletions. Patient 1's deletion also extends to 16q24.3 where it overlaps the coding region of ANKRD11, CDH15 and ZNF778. The coding regions of these three same genes are affected in Patients 22, 30 and 31, as well as three of the patients identified in DECIPHER. Excluding these deletions, there are 19 deletions that do not overlap genes previously associated with neurodevelopmental disorders. Overall, 14 of these deletions overlap in an interval (chr16 : 85 973 160–85 985 048; hg18) that includes the coding region of FBXO31 and MAP1LC3B (figure 2). These two same genes were affected in the 265-Kb deletion described by Butler et al (2012).9 Notably, neither FBXO31 nor MAP1LC3B is overlapped by a deletion in any of the 11 019 control cases examined.
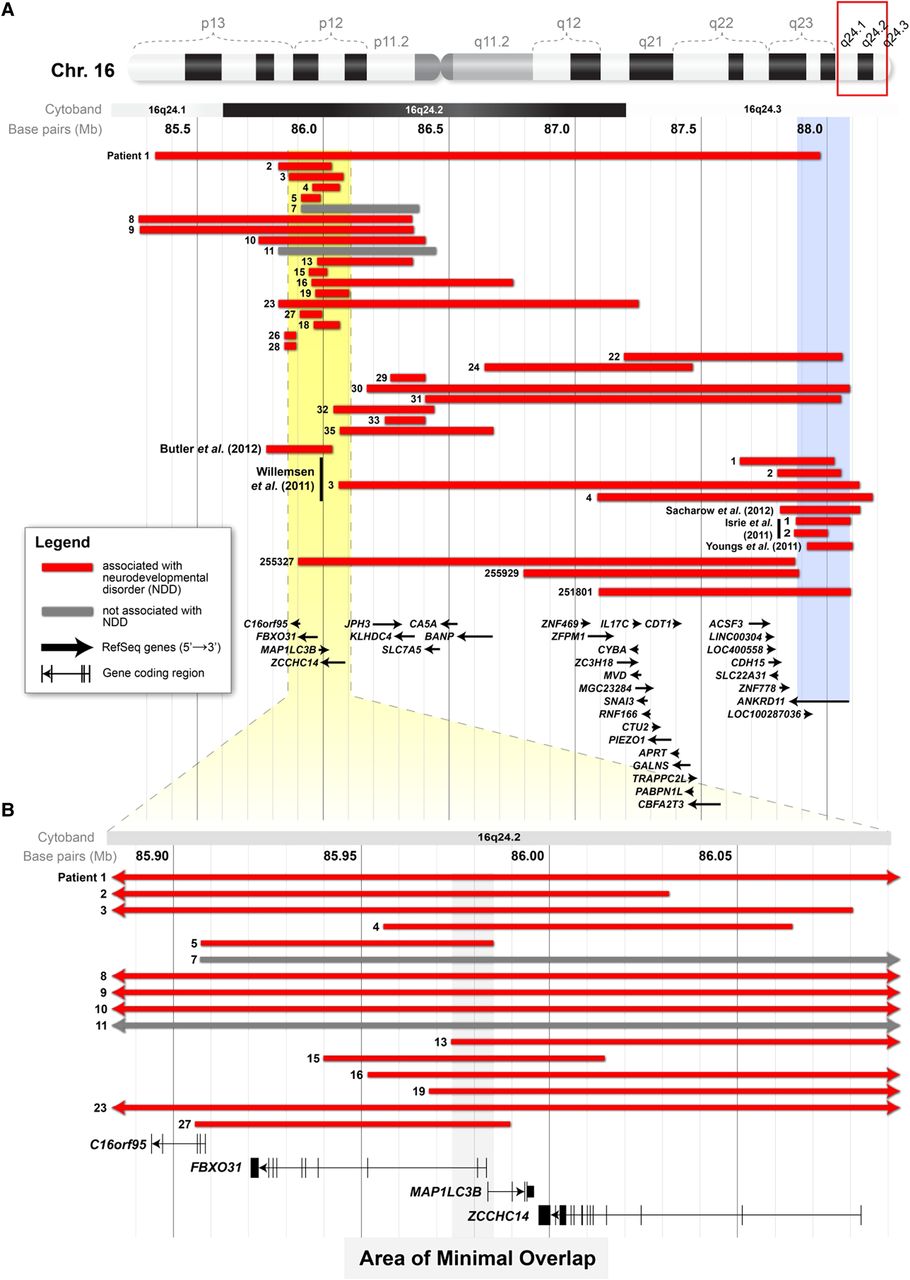
16q24.2 deletions associated with neurocognitive disease. (A) A schematic showing the interval of 85 163 271–88 279 649 (hg18) of chromosome 16. Displayed are the deletions from 19 individuals in this study with neurodevelopmental disorders (red bars), the deletions from two patients in this study demonstrated not to have the condition (grey bars) and deletions associated with neurodevelopmental disorders that have been reported elsewhere. The yellow area in A delineates the area (chr16 : 85 883 905–86 092 961; hg18) that is shown at higher magnification in B. The blue column shows the extent of the coding region of ANKRD11. (B) The area of minimal overlap (grey column) between the deletions in 14 patients in this study with neurodevelopmental disorders (red bars) and the deletion from the proband of Butler et al (2012).9 This area includes the coding regions of MAP1LC3B and FBXO31.
Patients 1, 6 and 7 in this study were found to have bilateral hydronephrosis on antenatal ultrasound. The deletion in Patient 6 extends from 16q24.1 to 16q24.2 and spans the entire coding region of FOXF1 (magenta column in figure 3), which is a candidate gene for congenital renal malformation and other abnormalities.8 The deletions in Patients 1, 6 and 7 overlap in a segment of 16q24.2 that includes the coding regions of four genes: C16orf95, FBXO31, MAP1LC3B and ZCCHC14 (figure 3). This region was also affected by the deletion described in Butler et al (2012)9 in an individual with bilateral vesicoureteral reflux among other congenital abnormalities (figure 3). There are 21 other patients in this study who bear deletions that affect some or all of the genes listed above. Of these, we can only rule out renal pathology in Patient 2 who underwent renal ultrasound (table 1, figure 3). Ultrasound data were not available for the other 19 patients (Patients 3–5, 8–13 15–20, 23, 27, 34), thereby preventing us from ruling out subclinical renal pathology in these individuals.

{kind=link}
{kind=link}
{kind=link}
16q24.2 deletions associated with congenital renal malformation. (A) A schematic showing the interval of 82 808 200–88 076 164 (hg18) of chromosome 16. This region is indicated by the area enclosed by the red box in the ideogram at the top. Three individuals in this study (Patients 1, 6 and 7), found to have bilateral hydronephrosis on antenatal ultrasound, bear deletions (shown in green) in 16q24.2 that overlap previously reported deletions from individuals with the same finding (Butler et al, 2012;9 Stankiewicz et al, 20098). The magenta column represents the extent of the coding region of FOXF1. (B) A magnified view of the interval highlighted in yellow in A. The area of minimal overlap (highlighted in grey) between the deletions in Patients 1 and 6 of this study and the proband of Butler et al (2012)9 includes the coding regions of C16orf95, FBXO31, MAP1LC3B and ZCCHC14. The area also overlaps two deletions from patients with bilateral pelviectasis, as reported by Stankiewicz et al (2009).8
Discussion
Here we report 35 individuals each with a deletion at 16q24.2. A total of 27 of these individuals were reported to have a neurodevelopmental disorder and/or the finding of bilateral hydronephrosis on antenatal ultrasound. Based on our findings, we propose that a subset of genes in 16q24.2 may regulate cognitive and renal development.
Novel candidate genes for neurodevelopmental disorders in 16q24.2
In all, 25 of the individuals in this study were reported to have ASD and/or intellectual disability. The deletions affecting this cohort are heterogeneous in terms of size and location within 16q24. Four of the individuals (Patients 1, 22, 30 and 31) have deletions that include the coding region of ANKRD11 (blue column in figure 2). We suggest that haploinsufficiency of this gene is the primary aetiology for ASD in these four individuals. Missense mutations in ANKRD11 are thought to cause KBG syndrome, which is characterised by ASD, intellectual disability, macrodontia, skeletal anomalies among other features.5,6,24 It is unclear whether any of the four patients has this condition, as there is insufficient clinical data in each case to make a diagnosis. We suggest that KBG syndrome is a likely diagnosis given that other individuals meeting the diagnostic criteria of the condition have also been found to have contiguous gene deletions affecting ANKRD11.4
The ANKRD11 locus is intact in the other 21 patients in this study with ASD or intellectual disability. This implies that there are other genes within 16q24 that, when haploinsufficient, may be associated with neurodevelopmental disorders. There is only one other report of a contiguous gene deletion in 16q24 that is associated with this condition.9 Butler et al (2012)9 described an individual with bilateral vesicoureteral reflux and cognitive impairment who had a 265-Kb deletion in 16q24.2. Reanalysis of this deleted segment demonstrates that it is indeed located in 16q24.2 and not in 16q24.3, as stated by the authors. This deletion affected the coding region of four genes: FBXO31, MAP1LC3B, C16orf95 and ZCCHC14. Butler et al9 suggested that haploinsufficiency of any of these genes may be associated with neurodevelopmental disorder.
Our data narrow this list of potential candidate genes for neurodevelopmental disorders from four to two. We report 14 individuals who have deletions that overlap the deletion identified by Butler et al (2012)9 in 16q24.2 and, at the same time, do not affect the coding region of ANKRD11 (figure 2). The area of minimal overlap of these 14 deletions includes the coding regions of FBXO31 and MAP1LC3B (grey column in figure 2). The first two exons of FBXO31 and the first exon of MAP1LC3B are included in the area of overlap. We suggest that one or both of these genes may be candidates for neurodevelopmental disorders. In support of this, we found that neither gene is overlapped by exonic deletions in any of the 11 019 control DNA samples that we inspected (see online supplementary table S3). In their study, Butler et al (2012)9 reasoned that MAP1LC3B was the most likely candidate for intellectual disability among the four. They based their argument on the gene's putative role in autophagy,25 which is associated with human neurodegenerative diseases, and the expression of the mouse homologue during development of the central nervous system in that animal.9 FBXO31, on the other hand, is a less likely candidate, as it has not been previously linked to neurocognitive development or disease (table 2). We suggest that both genes, FBXO31 and MAP1LC3B, should be investigated in terms of their function in cognitive development and disease.
Summary of the known information on the function and disease associations of the four candidate genes identified in this study
Patient 1's deletion spans 16q24.2q24.3 and includes the coding regions of all three candidate genes for neurocognitive disease discussed here (ie, MAP1LC3B, FBXO31, ANKRD11). It is impossible to determine the effect that haploinsufficiency of each of these genes may have on neurocognitive disease in this individual. We cannot rule out that there are still other candidate genes in 16q24.3q24.3 that may play a role. Two other genes that have previously been associated with neurocognitive disease and should be studied in more detail are CDH1526 and ZNF778,6 both found in 16q24.3 (figure 1).
Haploinsufficiency of 16q24.2 genes may be associated with congenital renal malformation
Three of the patients in this study, Patients 1, 6 and 7, were found to have bilateral hydronephrosis on antenatal ultrasound. Based on comparing our findings with previously reported cases, we suggest a separate aetiology for renal pathology in each patient.
Stankiewicz et al8 convincingly demonstrated that haploinsufficiency of FOXF1, a 16q24.1 gene, is associated with alveolar capillary dysplasia and the finding of bilateral hydronephrosis. The authors identified two mechanisms that may lead to haploinsufficiency of FOXF1: (1) disruption of the FOXF1 coding region or (2) deletion of cis regulatory elements that control expression of the gene. The latter mechanism is made more plausible by the recent identification of regulatory elements as far as 0.6 Mb from the coding region of another FOX gene, FOXG1.27 We suggest that haploinsufficiency of FOXF1 may contribute to the phenotype—particularly the renal abnormalities—of Patients 1 and 6. In support of this contention, the deletion in Patient 6 completely encompasses the coding region of FOXF1 (figure 2). In Patient 1, however, the FOXF1 locus is intact, and so mechanism 2 (ie, FOXF1 haploinsufficiency due to deletion of cis regulatory elements) is a more likely scenario. Whereas Patient 1's deletion is distal (225Kb) from the coding region of FOXF1, it overlaps several deletions reported by Stankiewicz et al (2009) (figure 3). The authors suggested that these deletions affect cis regulatory elements of FOXF1, perhaps altering its expression during kidney development and leading to renal malformation. We suggest that Patient 1's deletion has the same effect.
There is striking similarity between the clinical histories of Patients 1 and 6, particularly Patient 6, with the individuals described by Stankiewicz et al (2009).8 Six of the 10 patients reported by Stankiewicz et al (2009) were found to have bilateral hydronephrosis or pelviectasis on antenatal ultrasound. Eight of the patients succumbed to respiratory failure within the first month or two of life and demonstrated pulmonary hypertension. Similarly, Patient 6 died on the 16th postnatal day with respiratory failure and intractable pulmonary hypertension. In addition, Patient 6 demonstrated a septal heart defect, patent ductus arteriosus and vertebral abnormalities, three features seen in some of the cases in Stankiewicz et al (2009). Patient 1 also demonstrated heart and cardiac defects, but her respiratory distress at birth was transient, and she had gone on to survive to the age of 10 years at the time of this study. We propose that Patients 1 and 6 have a syndrome similar to the one described by Stankiewicz et al (2009) that is likely attributable to haploinsufficiency of FOXF1.
It is less likely that the renal pathology of Patient 7 is due to haploinsufficiency of FOXF1. This is because the deletion in this individual is quite distal (801 739 bp) from the coding region of FOXF1 (figure 3) and, furthermore, the medical history of the patient is not consistent with the syndrome described by Stankiewicz et al (2009).8 A more likely aetiology for renal pathology in Patient 7 is deletion of one of or some combination of C16orf95, ZCCHC14, FBXO31 and MAP1LC3B in 16q24.2 (figure 3). Butler et al (2012)9 suggested a similar aetiology for the renal malformation seen in their patient, who had a deletion affecting these four same genes. Our finding that Patient 2 had a deletion affecting the same four genes (figure 3), but no signs of renal pathology, as determined by ultrasound, implies that there may be reduced penetrance or variable expressivity of the renal phenotype among patients with deletions affecting the four genes.
The common involvement of C16orf95, ZCCHC14, FBXO31 and MAP1LC3B in two unrelated cases of bilateral hydronephrosis raises the possibility that haploinsufficiency of any one (or some combination) of the genes may be associated with congenital renal malformation. At present, only one of the genes, ZCCHC14, has been shown by in situ hybridisation to be expressed in the developing urinary tract of the mouse (table 2). None of the genes has been associated with renal malformation in any other study (table 2). Further data on the developmental expression and function of each gene as well as the human disease association of intragenic mutations within each gene would help clarify which of the four could be a candidate for congenital renal malformation.
In this study, we showed that deletions in chromosome region 16q24.2 are associated with neurodevelopmental disorders, such as intellectual disability and ASD, and congenital renal malformation. We suggest that the most likely candidate genes for these conditions are C16orf95, ZCCHC14, MAP1LC3B and FBXO31. The function of each of these genes in development and disease should be studied in more detail.
Note added in proof
While this work was in revision, Szafranski et al (PMID: 23034409) published a report showing that deletions within a cis regulatory region located 250 Kb upstream of the FOXF1 locus in 16q24.1 can cause alveolar capillary dysplasia. This region encodes long, non-coding RNAs, which the authors posit are involved in the long-range regulation of FOXF1. The deletion in Patient 6 of this study overlaps this region of interest.
Acknowledgments
The authors would like to thank the patients who contributed to this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors Conceived the study: GRH, ACL, CRM, NDR, SWS and JEC. Wrote the manuscript: GRH, DC, ACL and NDR. Provided clinical data: DC, MP, SD, LFE, BAF, JCS, JAR, LGS, AKV, MG, JCH, RB, DJS and VY. Analysed/interpreted data: GRH, DC, ACL, MP, CRM, JAR, LGS, AKV, MG, JCH, DJS, SWS and NDR. Revised manuscript critically: all authors. Approved submitted version of manuscript: all authors.
-
Funding This work was supported by grants awarded by the Canadian Institute of Health Research, the Kidney Foundation of Canada and the Canada Research Chair Program (to NDR). The Centre for Applied Genomics (TCAG), McLaughlin Centre and the Technology Innovation Centre are funded by Genome Canada and the Ontario Genomics Institute (SWS). GRH was supported by a research scholarship from the Comprehensive Research Experience for Medical Students (CREMS) Program, Faculty of Medicine, University of Toronto. SWS holds the GlaxoSmithKline-CIHR chair in Genome Sciences at the University of Toronto and the Hospital for Sick Children.
-
Competing interests At the time of this study, JAR and LGS were employees of Signature Genomics, a subsidiary of PerkinElmer.
-
Patient consent Obtained.
-
Ethics approval Research Ethics Board, The Hospital for Sick Children.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Detailed microarray/aCGH data are available upon request.
Linked Articles
- Correction